
howitreat.in
A user-friendly, frequently updated reference guide that aligns with international guidelines and protocols.
Alpha Thalassemia
Introduction:
- It is a group of hereditary anemiascharacterized by reduced or absent production of alpha chains of hemoglobin.
Epidemiology:
- Common in Africa, Mediterranean countries, middle east and south east Asia.
Etiology:
- Autosomal co-dominant inheritance.
- Deletion of genes coding for α chain of hemoglobin.
- This gene is located on short arm of chromosome 16.
- Unlike β & δ which are single for haploid genotype, α is duplicated on chromosome 16.
- As both HbA and HbF contain α chains, in α thalassemia both fetus and adults are affected.
Pathogenesis:
- In newborn, excess of γ chains aggregate to form γ4 tetramers, which are known as Hb Barts. Hb Barts has very high oxygen affinity, which results in severe tissue anoxia and intrauterine death.
- In adults, excess of β chains also aggregate to form β 4 tetramer, which is known as HbH. Oxygen affinity of HbH is 10 times that of HbA due to lack of heme-heme interaction and absence of Bohr effect. Oxygen dissociation curve is a rectangular parabola shifted to extreme left.
- HbH is unstable and precipitates causing hemolytic anemia.
Types α thalassemia
Genotype | Phenotype | Hematological findings | Severity | Hemoglobin patterns |
-α/ αα | Silent carrier | Normal or slight reticulocytosis | Normal | Normal |
-α/ -α | α thalassemia trait | Mild anemia, microcytic hypochromic RBCs, target cells, basophilic stippling, poikilocytosis | Mild to moderate | Newborns- HbBarts Adults- Normal or some HbH |
--/ αα | ||||
--/ -α | Hemoglobin H disease | Moderate to marked anemia, microcytic hypochromic RBCs, target cells, basophilic stippling, poikilocytosis | Chronic, moderately severe hemolytic anemia, Splenomegaly | Newborns- HbBarts Adults- HbH |
--/ -- | Hydropsfetalis with Hemoglobin Barts | Marked anemia, macrocytic hypochromic RBCs, marked Anisopoikilocytosis, numerous NRBC | Fatal | HbBarts Hb Portland |
Investigations:
- Hemogram:
- Hemoglobin- Reduced.
- Blood indices-MCV and MCH are disproportionately low when compared with severity of anemia.
- Microcytic hypochromic RBCs
- Many targets cells are seen.
- Acanthocytosis.
- Basophilic stippling present.
- RBCs are pitted several times and they look like surface of golf ball.
- Reticulocyte count- Increased.
- Hemoglobin electrophoresis on cellulose acetate at pH 8.6- Both HbH and HbBarts are fast moving hemoglobins.
- Demonstration of HbH inclusions: Done by incubating blood with solution of redox dye-Ex –Brilliant cresyl blue.
- Globin chain synthesis rate studies- Done by using radioactive amino acids.
- Gene analysis study
- Antenatal diagnosis can be done by analyzing the cells obtained through amniocentesis and chorionic villi biopsy
Treatment: Only HbH disease must be treated. Treatment is similar to beta thalassemia major/ intermedia
- Chronic hypertransfusion.
- Splenectomy.
- Folic acid supplements.
- Treatment of endocrine deficiencies occurring due to hemochromatosis.
- Avoid oxidant injury.
Related Disorders:
- Hemoglobin constant spring (HbCS)
- Elongated α chain variant of HbH
- Combination of two structurally abnormal α chains, each elongated by 31 amino acids at the carboxy terminal end, and two normal β chains.
- It occurs due to mutation of chain termination codon. (UAA changes to CAA which codes for glutamine, transcription of mRNA continues till next stop codon is reached)
- Abnormal α chains are ineffectively synthesized due to reduced stability of mRNA translocation apparatus. This leads to overall deficiency of α chains.
- These patients present with mild anemia.
- α Thalassemia –mental retardation syndrome.
- 2 forms
- Mutation of ATR-16- Gene on chromosome 16
- Mutation of ART-16-Gene in X chromosome.
- Associated with widespread dysmorphic features
- 2 forms
- α -Thalassemia with myelodysplasia (Acquired alpha thalassemia).
- Seen in patients with MDS or AML.
- Associated mutation of ATR-X gene which leads to inactivation of alpha genes in neoplastic hematopoietic cells.
- Have dimorphic anemia with RBCs containing HbH.
Figures:
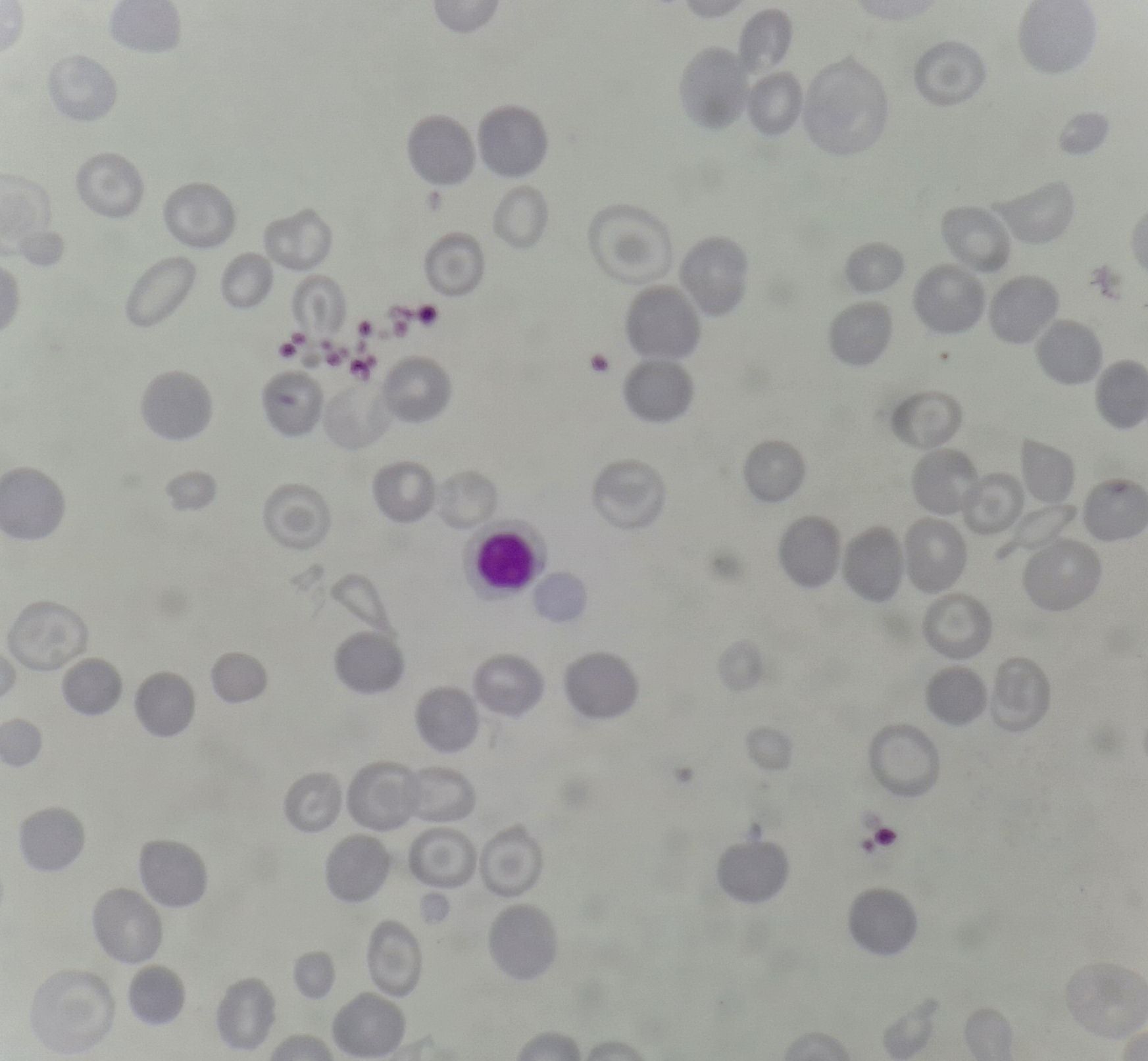
Figure 8.13.1- Alpha thalassemia trait- Peripheral smear
An Initiative of
Veenadhare Edutech Private Limited
1299, 2nd Floor, Shanta Nivas,
Beside Hotel Swan Inn, Off J.M.Road, Shivajinagar
Pune - 411005
Maharashtra – India

howitreat.in
CIN: U85190PN2022PTC210569
Email: admin@howitreat.in
Disclaimer: Information provided on this website is only for medical education purposes and not intended as medical advice. Although authors have made every effort to provide up-to-date information, the recommendations should not be considered standard of care. Responsibility for patient care resides with the doctors on the basis of their professional license, experience, and knowledge of the individual patient. For full prescribing information, including indications, contraindications, warnings, precautions, and adverse effects, please refer to the approved product label. Neither the authors nor publisher shall be liable or responsible for any loss or adverse effects allegedly arising from any information or suggestion on this website. This website is written for use of healthcare professionals only; hence person other than healthcare workers is advised to refrain from reading the content of this website.