
howitreat.in
A user-friendly, frequently updated reference guide that aligns with international guidelines and protocols.
Auto-immune Hemolytic Anemia
Introduction:
- It is a hemolytic anemia caused due to antibodies against basic component of Rh system (70%) and rarely against antibodies against other systems.
Epidemiology:
- Warm antibody AIHA (65% of AIHA)
- 5-10/1million population/ year
- Usually seen after 40 years of age. Peak in 7th decade.
- Cold agglutinin disease (30% of AIHA)
- 1.4/ million population/ year
- Common in women
Etiology:
- Warm antibody type (IgG type antibody which is active at 37oC. Cause extravascular hemolysis in spleen)
- Primary or idiopathic (50% cases)
- Leukemia and lymphoma- CLL, low grade B cell NHL, Hodgkin disease, AML, multiple myeloma, etc.
- Neoplasms like ovarian cysts and carcinomas, thymoma, Kaposi’s sarcoma etc
- Autoimmune disorders- SLE, rheumatoid arthritis, Sjogren syndrome, ulcerative colitis, scleroderma, antiphospholipid antibodies etc
- Infections- EBV, HCV, HIV etc
- Drugs-
- Drug dependent subtype- Antibodies are formed in response to neoantigen formed by binding of a drug to cell surface antigen.
- Drug independent subtype- Offending drug induces an autoimmune response that persists even in absence of the drug.
- Drugs include: Ceftriaxone and other cephalosporines, piperacillin, penicillin, quinidine, methyl dopa, L dopa, procainamide, mefenamic acid, Interferon alfa, Purine analogues, Chlorambucil, lenalidomide, bendamustine, PD1 inhibitors etc
- Immunodeficiencies- Common variable immunodeficiency, ALPS, Wiskott Aldrich syndrome
- Cold agglutinin type (IgM type antibody, which is active at 0-4oC. Cause intravascular hemolysis usually in liver. Hemolysis occurs due to classical complement pathway. Along with hemolytic anemia, they also have acrocyanosis/ Reynaud’s phenomenon as RBCs agglutinate in acral portions which are cooler. It occurs due to stasis of peripheral circulation secondary to red cell agglutination)
- Idiopathic
- Anti I antibodies- They react strongly with adult RBCs
- Anti i antibodies- they react strongly with cord RBCs
- Infections such as mycoplasma, EBV, CMV, SARS-CoV-2
- Low grade NHL (Lymphoplasmacytic lymphoma, marginal zone lymphoma) and aggressive NHL.
- Aggressive B Cell lymphoma
- Drugs: Etanercept, Eculizumab
- Idiopathic
- Cold hemolysins (Paroxysmal cold hemoglobinuria). Donath Landsteiner antibody (IgG type anti P antibody) is often present which binds to RBC at low temperature and causes complement mediated lysis of cell when temperature is raised to 37oC. DAT is positive to C3 only. Presents with fever, leg pain, abdominal/back pain and hemoglobinuria. Seen very rarely now)
- Primary
- Infections- Adenovirus, Influenza A, syphilis, CMV, infectious mononucleosis, varicella zoster virus, measles, mumps, mycoplasma
Pathogenesis:
- Warm antibody
- Cell mediated immune destruction
RBCs coated with IgG antibodies get bound to splenic macrophages/ Kupffer cells in liver
↓
Part of coated membrane is lost(pitting) and remaining membrane seals itself.
OR
Whole RBC may be engulfed
↓
Hemolytic anemia and acquired spherocytosis
- Complement mediated intravascular hemolysis
IgG along with C3 on surface of RBC promote cell leukocyte interaction
↓
More severe hemolysis
- Polymorphism of TLA-4 gene
↓
Loss of immune tolerance due to defective action of CTLA-4 on regulatory T cells
- Cold antibody
Binding of IgM with erythrocyte occurs at cold temperature (occurs in extremities)
↓
Complement gets attached to this antigen antibody complex which occurs at temperature of 20-25oC.
↓
Upon warming antibody is detached from the cell, but the complement remains on the surface and gets activated
↓
Direct cell lysis leading to intravascular hemolysis
Factors that affect the rate of hemolysis in immune hemolytic anemias
- Class of immunoglobulin coating the erythrocytes. For the IgG subclasses, the affinity of macrophage receptors and rate of hemolysis is greatest for IgG3 and IgG1.
- The number of Ig molecules per erythrocyte. High density antigens bind more Ig per cell than low density antigens.
- Ability of Ig to activate complement. IgM and IgG (IgG1 and IgG3) can activate complement. IgG2 is less efficient and IgG4 is unreactive with the complement.
- Thermal amplitude of the antibody. Warm (37oC) reacting antibodies may cause hemolysis but cold (0-4oC) reacting antibody does not.
- The activity of macrophages- i.e. their ability to sequester sensitized cells.
Other factors contributing to anemia
- Decreased marrow function due to
- Action of auto antibodies against reticulocytes and erythroblasts
- Associated folate deficiency due to increased demand
- Infiltration of lymphoproliferative disease in bone marrow
- In case of SLE, in spite of presence of autoantibodies there may very less hemolysis due to
- Reticulo endothelial blockade- Occurs in process of removal of immune complexes by RES
- Hypocomplementemia due chronic activation of complement system.
Clinical Features:
- Sudden onset of very severe anemia
- Jaundice
- Splenomegaly
- High risk of thrombosis (15-20% of patients with AIHA have thrombosis)
Investigations:
- Hemogram
- Hemoglobin content- Reduced
- Hematocrit- Reduced
- Normocytic normochromic RBCs
- Many micro-spherocytes are seen- Consistent and diagnostically important feature
- Schistocytes and many other poikilocytes also may be noted
- Many polychromatophilic cells and nucleated RBCs are noted
- Erythrophagocytosis by monocytes is sometimes seen
- Gross hemagglutination may be noted in case of cold hemagglutinin disease
- Mild leukocytosis and neutrophilia
- Bone marrow examination
- Erythroid hyperplasia
- Erythrophagocytosis by macrophages
- Underlying LPD may be noted in some cases
- Aplastic crisis may be seen when patient contracts certain viral infections
- Reticulocyte count- Elevated (Causes of reticulocytopenia include- Acute phase of AIHA, presence of autoantibodies against erythrocyte precursors, Marrow infiltration, hematinic deficiency, aplastic anemia, Parvovirus B19 infection)
- Bilirubin- Increased unconjugatedbilirubin
- Coomb’s test (Antiglobulin test)
- Coomb’s reagent is rabbit IgM antibody raised against human IgG
- Direct Coomb’s test
- Patient’s saline washed RBCs are added to Coomb’s reagent
- Agglutination indicates presence of antibodies over the surface of RBCs
- Indirect Coomb’s test
- Patient’s serum is added to a panel of “O” blood group RBCs and incubated
- Upon addition of Coomb’s reagent, if agglutination occurs it indicates presence of free antibodies in patients serum
- Negative Coomb’s test in AIHA (Coomb's negative AIHA. 3-10% of AIHA are Coomb’s negative)
- Insufficient number of antibodies(Coomb’s test detects antibodies if their concentration is minimum of 100-500 molecules per cell. In vivo removal of sensitized cells by macrophages occurs at much lower concentration)
- Low affinity antibodies
- Immunoglobulin which is not tested for Ex: IgA only AIHA
- Positive Coomb’s test with no evidence of hemolysis
- Individual’s macrophages may not be active in removing sensitized cells
- Patients with hypergammaglobulinemia (Liver disease, Chronic infections, SLE, paraproteinemia, IVIg therapy and use of Daratumomab)- Coomb’s test may be positive because of nonspecific binding of Ig to erythrocytes
- Thermal amplitude of antibodies may be less than 37oC
- Drugs like alpha methyl Dopa, IVIg, ATG
- Complements on erythrocytes- Increased C3d is found in very ill patients
- If ICT is positive and DCT is negative, it indicates presence of allo antibody stimulated by prior transfusion or pregnancy.
- MonospecificCoomb's test
- IgG alone- Warm antibody AIHA, Drug related immune hemolytic anemia
- Complement alone (C3d)- Warm antibody AIHA with subthresholdIgG deposition, Cold agglutinin disease, Paroxysmal cold hemoglobinuria, Drug induced AIHA
- Both positive- Warm antibody AIHA, Drug induced (rarely)
- Reticulocyte production index- 6-7
- Specific test for cold agglutinin (Ehrlich finger test)
- It is used for demonstrating hemolysis in microcirculation at cold temperature
- Venous blood flow in two fingers is stopped with rubber band
- One finger is immersed in cold water (20oC) and other finger in warm water(37oC)
- Using capillary tubes, blood is taken from each finger and tubes are centrifuged in a microhematocrit centrifuge.
- Plasma layer is examined for hemolysis
- Positive test is indicated by hemolysis in the blood from the finger incubated in cold water
Diagnostic criteria for the diagnosis of cold agglutinin syndrome
Essential:
- Chronic haemolysis - high indirect bilirubin, low haptoglobin, high LDH, and (often) high absolute reticulocyte count
- Coomb’s strongly positive for C3d; negative for IgG
- Cold agglutinin titre > 64 at 4 °C (specimen must be kept at 37-38 °C from sampling until plasma/serum has been removed from the cells/clot)
- No overt malignant disease or relevant infection - clinical assessment for malignancy; radiology as required; exclude recent infection with Mycoplasma or EBV
- Evidence of a clonal B-cell disorder - as assessed by electrophoresis, flow cytometry, and/or bone marrow biopsy
Desirable:
- Monoclonal IgM/kappa in plasma/serum (or, rarely, IgG or lambda phenotype); the specimen must be kept at 37-38 °C from sampling until plasma/serum has been removed from the cells/clot
- B-cell kappa-to-lambda ratio > 3.5 (or, rarely, < 0.9) - as assessed by flow cytometry in bone marrow aspirate
- Cold agglutinin-associated lymphoproliferative disorder by histopathology (on bone marrow biopsy)
- MYD88 p.L265P mutation not found - as assessed in bone marrow
Prognosis:
- Adults- Remitting, relapsing course
- Children- AIHA found after infection has self-limiting course
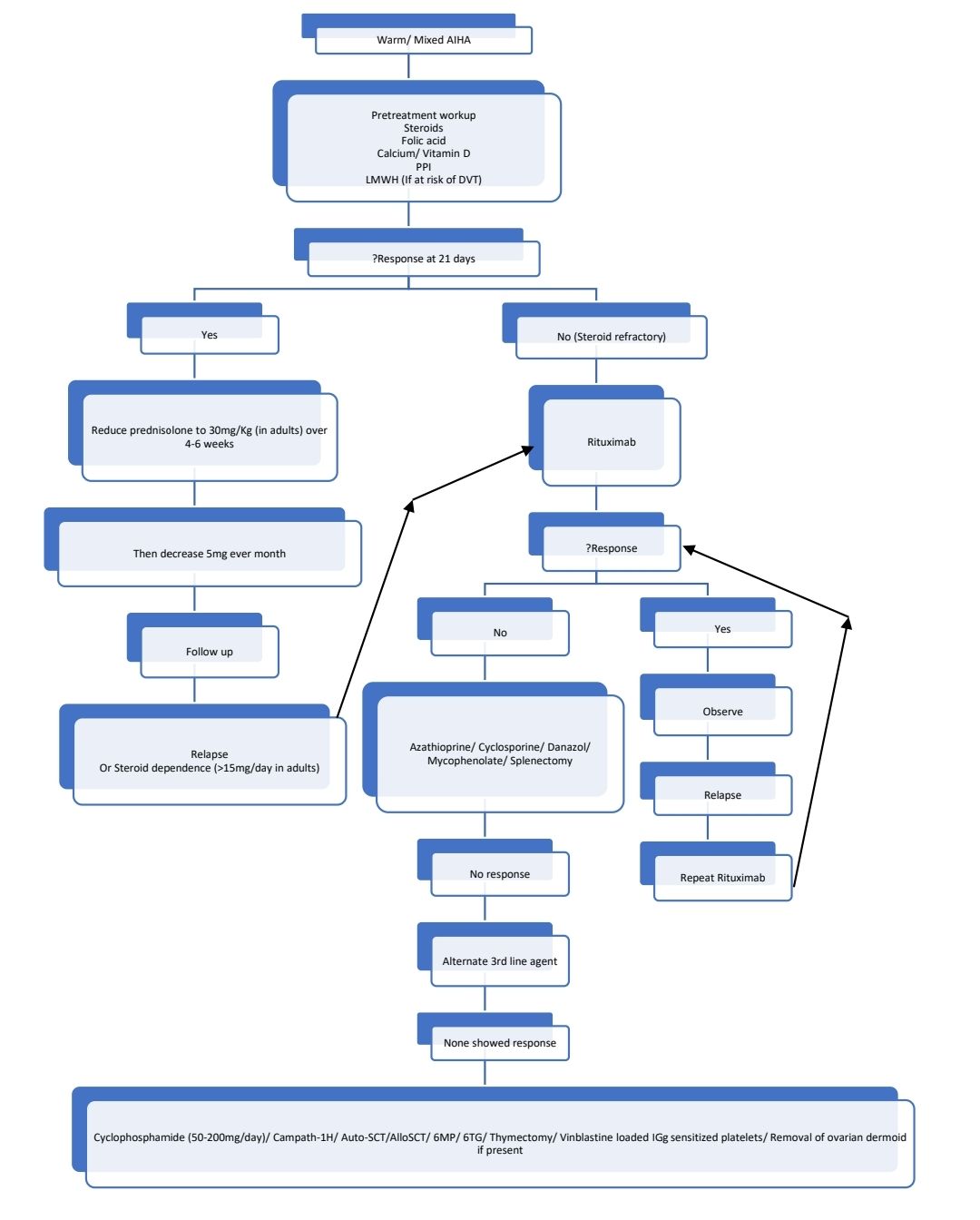
Indications for Treatment:
- Severe decompensated hemolysis (If hemolysis is compensated, there is no need for treatment)
Pretreatment Work-up:
?Hemolytic anemia (Yes- if there is anemia, reticulocytosis, indirect hyperbilirubinemia, raised LDH and decreased haptoglobin)
↓
?Autoimmune hemolytic anemia (Yes- If DCT is positive and other causes of DCT positive hemolytic anemia are rule out such as immediate/delayed hemolytic transfusion reaction, Post BMT hemolysis due to major ABO mismatch/ passenger lymphocyte syndrome, Hemolytic disease of newborn and other causes of hemolysis with incidental DCT positivity)
↓
?Warm/ cold AIHA (Do- Monospecific DCT with antibodies to IgG and C3d)
↓
?Primary/ Secondary (Tests such as serology, CT scan, BM aspiration etc)
- History
- Examination
- Hemoglobin
- TLC, DLC
- Platelet count
- Peripheral smear
- DCT
- Monnspecific DCT
- Reticulocyte count
- Urine dipstick &Micro
- LFT: Bili- T/D SGPT: SGOT: Albumin: Globulin:
- Creatinine
- LDH
- Haptoglobin
- HIV:
- HBsAg:
- HCV:
- SPEP/ Immunofixation electrophoresis
- ANA, Anti DsDNA
- S. Immunoglobulins: IgG: IgG: IgM:
- CT C/A/P
- BMA and Bx (Done in case of Cold agglutinin disease, Age >60 years, suspicion BM involvement)
- Peripheral T Subset analysis (Especially in children)
- Cold agglutinin thermal amplitude
Treatment plan:
Severe AIHA
- Blood transfusion with least incompatible blood
- IVIg- 0.4gm/Kg/day for 5 days
- Consider high dose methyl prednisolone- 100-200mg/day (Carries high risk of infection)
- Emergency splenectomy (For relapsed/ refractory cases)- Do vaccination 14 days after splenectomy
Drug induced AIHA
- Stop offending drug
- Supportive care
Paroxysmal cold hemoglobinuria
- Supportive care (Keep warm, folic acid, transfusions)
- Steroids- If severe/ persistent disease
About Each Modality of Treatment:
- Secondary AIHA: Treatment of cause
- T/B NHL- Chemotherapy
- Hodgkin's disease- Chemotherapy
- Solid tumors- Surgery and steroids
- Ovarian dermoid cyst- Surgical excision
- SLE- Steroids with Azathioprine/ MMF
- Ulcerative colitis- Steroids/Azathioprine
- CVID- IVIg replacement along with steroids. Rituximab is the second line agent.
- Autoimmune lymphoproliferative disorder- Steroids. If no response- MMF/Sirolimus
- WiskottAlldrich syndrome- Steroids. If no response- BMT.
- Mycoplasma induced cold agglutinin disease- Appropriate antibiotics
- Infectious mononucleosis- If hemolysis is mild, monitor. If hemolysis is severe use steroids.
- HCV- Hepatitis C eradication
- Allogeneic SCT- Steroids. If no response Rituximab. If no response- Splenectomy or T cell infusion.
- Organ transplantation- Reduction of immunosuppression and steroids
- Drug induced- Discontinue offending drug. Steroids are generally not required, but can be used if there is severe persistent hemolysis.
- Steroids:
- Prednisolone- 1-2mg/kg/day in divided doses
- Helpful in 2/3rd of patients
- 20% achieve complete remission
- They decrease antibody production and also decrease sequestration by macrophages.
- Improvement in hemoglobin concentration occurs after 3-4 weeks.
- Start tapering the dose once hemoglobin is >10gm/dL or after maximum of 3 weeks.
- Reduce the dose to 30mg/day over 4-6 weeks. Then decrease the dose by 5mg every month.
- There is high risk of relapse if steroids are stopped within 6 months.
- Proton pump inhibitors may be added to avoid gastric erosion
- Some may need maintenance dose of 10-15mg on long term basis, which can be given, as such doses rarely lead to any side effects.
- Continue therapy till DCT becomes negative.
- Add calcium/ Vitamin D and bisphosphonates for patients who need long term steroids
- Avoid giving high dose methylprednisolone/ dexamethasone, in view of high risk of serious infections
- Blood transfusion
- Should be avoided as far as possible, as it leads to more hemolysis.
- Should be given in case of acute hemolytic crisis
- If anemia is life threatening, give least incompatible blood and transfusion should be done very slowly, with close watch on any hemolytic transfusion reactions.
- Use warmer if patient has cold agglutinin disease
- Folic acid- 5mg-OD
- VTE prophylaxis
- Risk of VTE is 21%
- Give low molecular heparin
- Rituximab
- 40-100% response rate
- Dose- 375mg/m2 – IV- weekly for 4 weeks
- Low dose i.e. 100mg- weekly for 4 weeks is also found to be effective.
- Removes B cells which produce autoantibodies
- Indications
- Failure to respond to steroids after 3 weeks of trial
- Relapse during steroid reduction
- Dependence on prednisolonei.e- requirement of >15mg/day prednisolone to maintain hemoglobin
- Intractable side effects of steroids
- Azathioprine
- 80mg/m2/day- for 3-4 months
- 60% response rate
- Cyclosporine
- 5mg/kg/day in 2 divided doses
- Maintain trough levels between 200-400ng/ml
- Mycophenolatemofetil- 500mg- BD/TID
- Danazol- 600-800mg/day
- Splenectomy
- Results of splenectomy
- 1/3rd- Achieve complete remission and do not require steroids
- 1/3rd- Significantly reduced steroid requirement
- 1/3rd- No or only transient response, as RBC lysis continues in Kupffer cells in liver
- Incidence of postoperative VTE is common. Hence thromboprophylaxis is must.
- Results of splenectomy
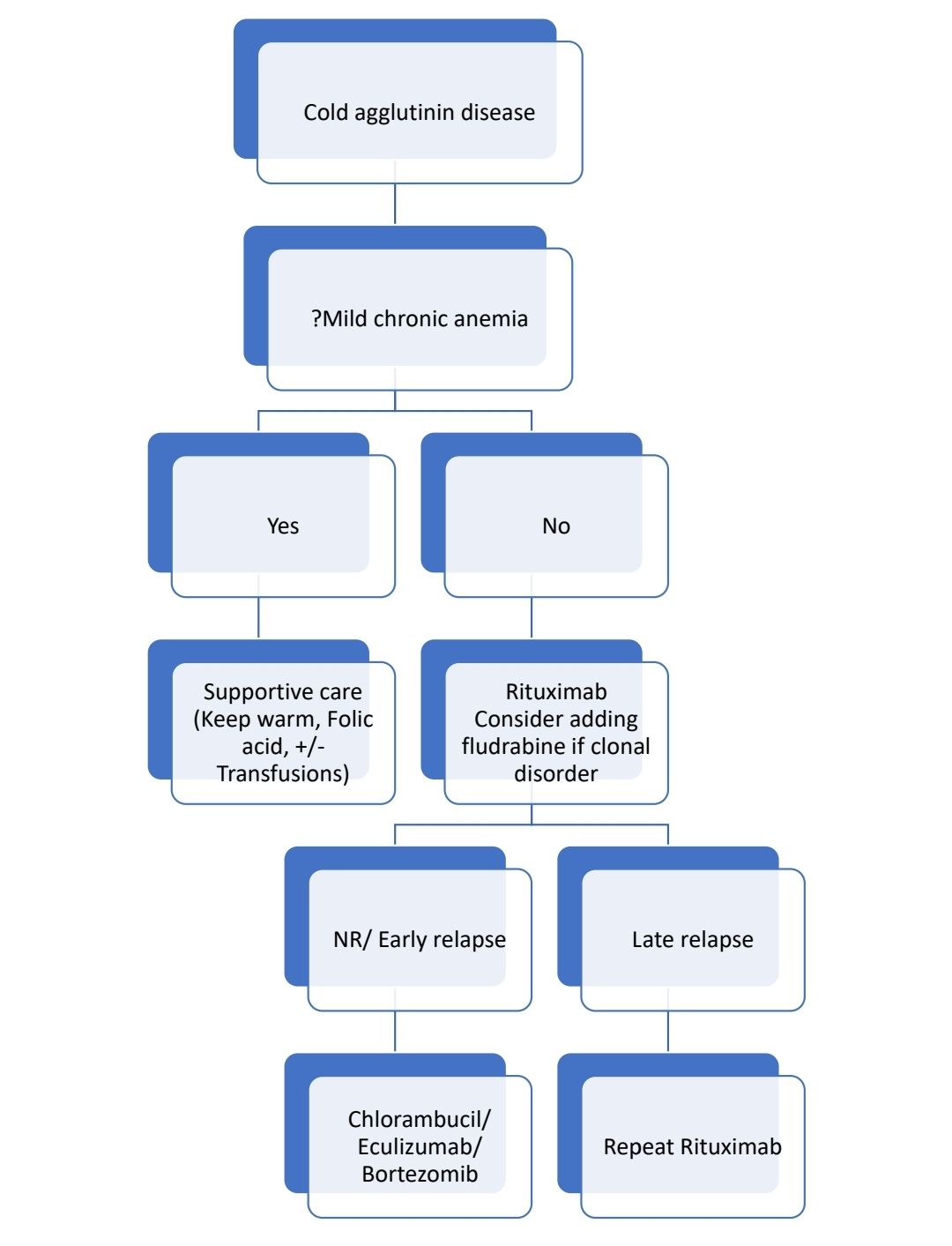
Treatment of cold agglutinin disease
- Avoid exposure to cold and use electrically heated gloves and socks
- Rituximab- Same as above
- Chlorambucil-
- Dose- 10mg/kg- for 14days, every 4 weeksOr continuous 2-4 mg/day
- Long term treatment can cause MDS and AML
- Steroids- Not very useful
- Splenectomy- Not useful
- Blood transfusion through inline blood warmer
- Plasma exchange- to reduce the titer cold agglutinin
Other treatment options:
- Immunoproteasome inhibitors:
- Bortezomib
- KZR-616
- FcRn inhibition: Efgartigimod
- Inhibitors of phagocytosis
- Syk kinase inhibition: Fostamatinib
- Bruton kinase inhibition: Rilzabrutinib
Special Situations:
- Childhood AIHA
- 77% are self limiting, requiring only short term treatment
- Warm type is common
- Needs testing for ALPS and primary immunodeficiency
- Unusually high association with Giant cell hepatitis
- Treatment- Similar to adults
- AIHA in pregnancy
- 1 in 1,40,000 pregnancies
- Associated with higher risk of preeclampsia
- Treated with prednisolone
- If no response, Azathioprine is acceptable in pregnancy
- Splenectomy can be considered in second trimester
- Low dose aspirin must be given to prevent preeclampsia
- Serial USG and doppler of fetal middle cerebral artery is necessary to assess fetal anemia
- Give antenatal and 6 weeks post-partum thromboprophylaxis
- Test neonate for evidence of hemolysis and DCT. Management of hemolysis is similar to hemolytic disease of newborn.
Related Disorders:
- Evan’s syndrome
- Autoimmune hemolytic anemia is associated with immune thrombocytopenia
- There is higher incidence of following underlying illnesses
- Children- immunodeficiency, autoimmune lymphoproliferative disease
- Adults- SLE, T cell lymphoma, APLA
- Prognosis is poor compared to primary warm AIHA.
- Treatment is similar to AIHA
- Benign cold agglutinins
- They are called benign because their thermal amplitude and concentration are not high enough to cause clinical problems.
Characteristic of agglutinin | Pathological agglutinins | Benign agglutinins |
Antibody class | IgM | IgM |
Antibody specificity | Usually anti-I but secondary CHD may be anti-i | Anti-I |
Antibody clonality | Monoclonal in idiopathic type: Polyclonal in secondary type | Polyclonal |
Thermal amplitude | 0-31oC | 0-4oC |
Agglutination at room temperature | Significant | Not present |
Agglutination in albumin | Enhanced | No effect |
Titer | Usually >1:1000 | <1:64 |
Coomb’s test | positive with polyspecific AHG and monospecific anti-complement | Negative |
Figures:
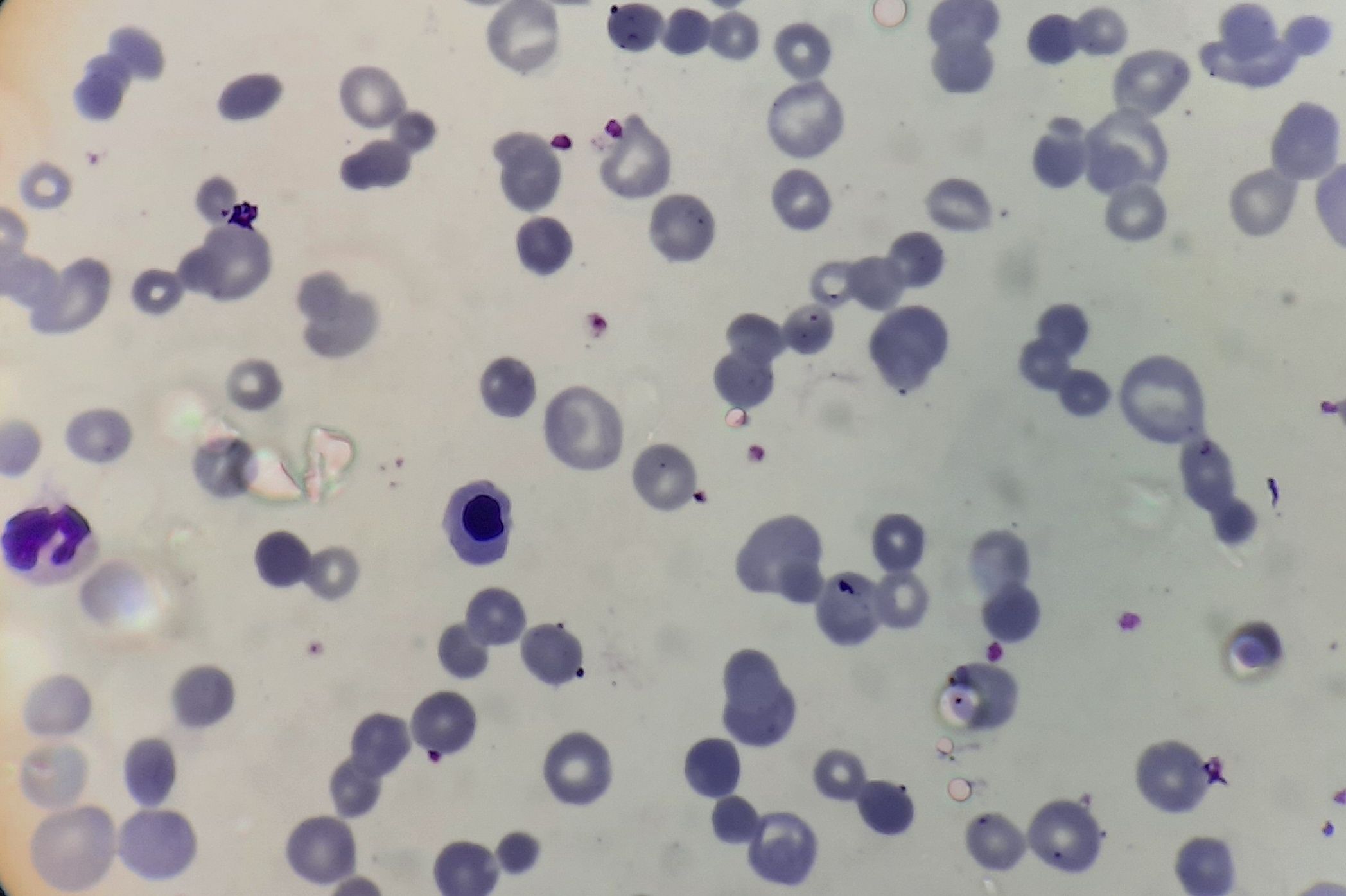
Figure 8.21.1- Autoimmune hemolytic anemia- Warm antibody- Peripheral smear
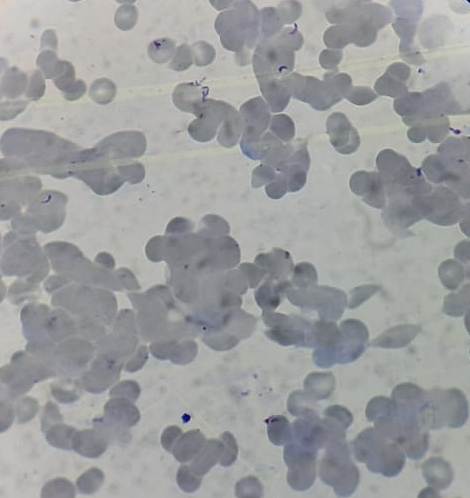
Figure 8.21.2- Autoimmune hemolytic anemia- Cold agglutinin disease- Peripheral smear
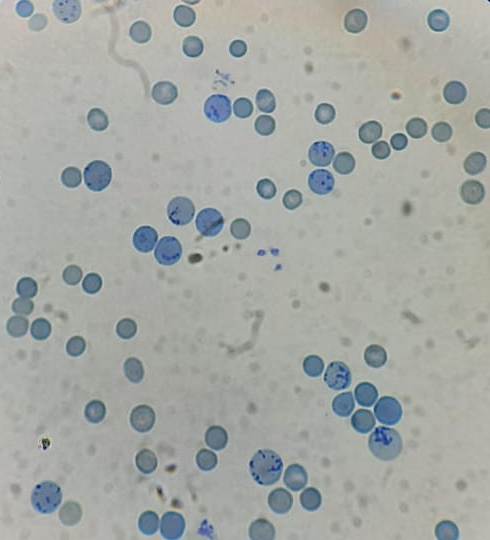
Figure 8.21.3- Autoimmune hemolytic anemia- Increased reticulocyte count
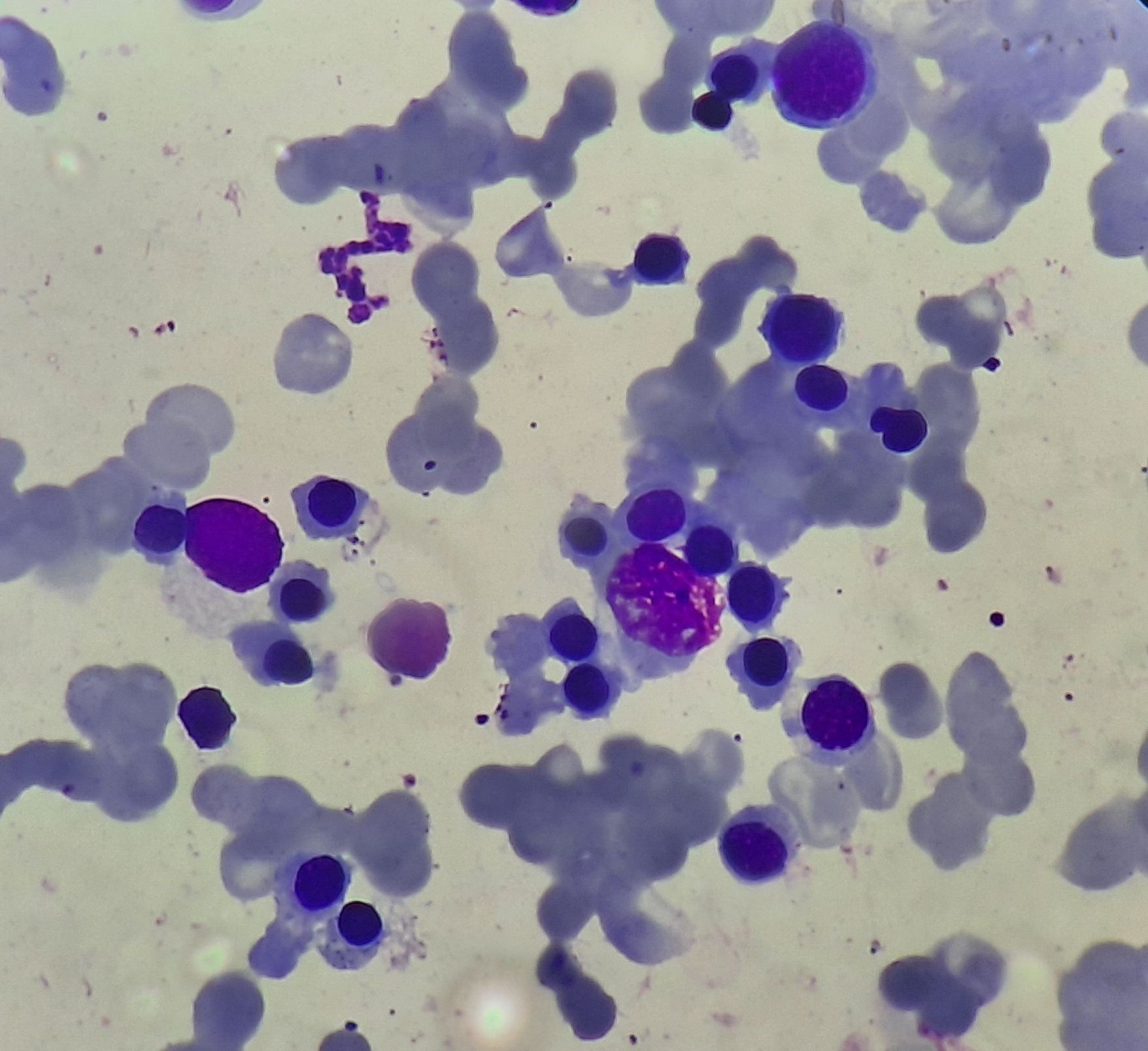
Figure 8.21.4- Autoimmune hemolytic anemia- Bone marrow aspiration
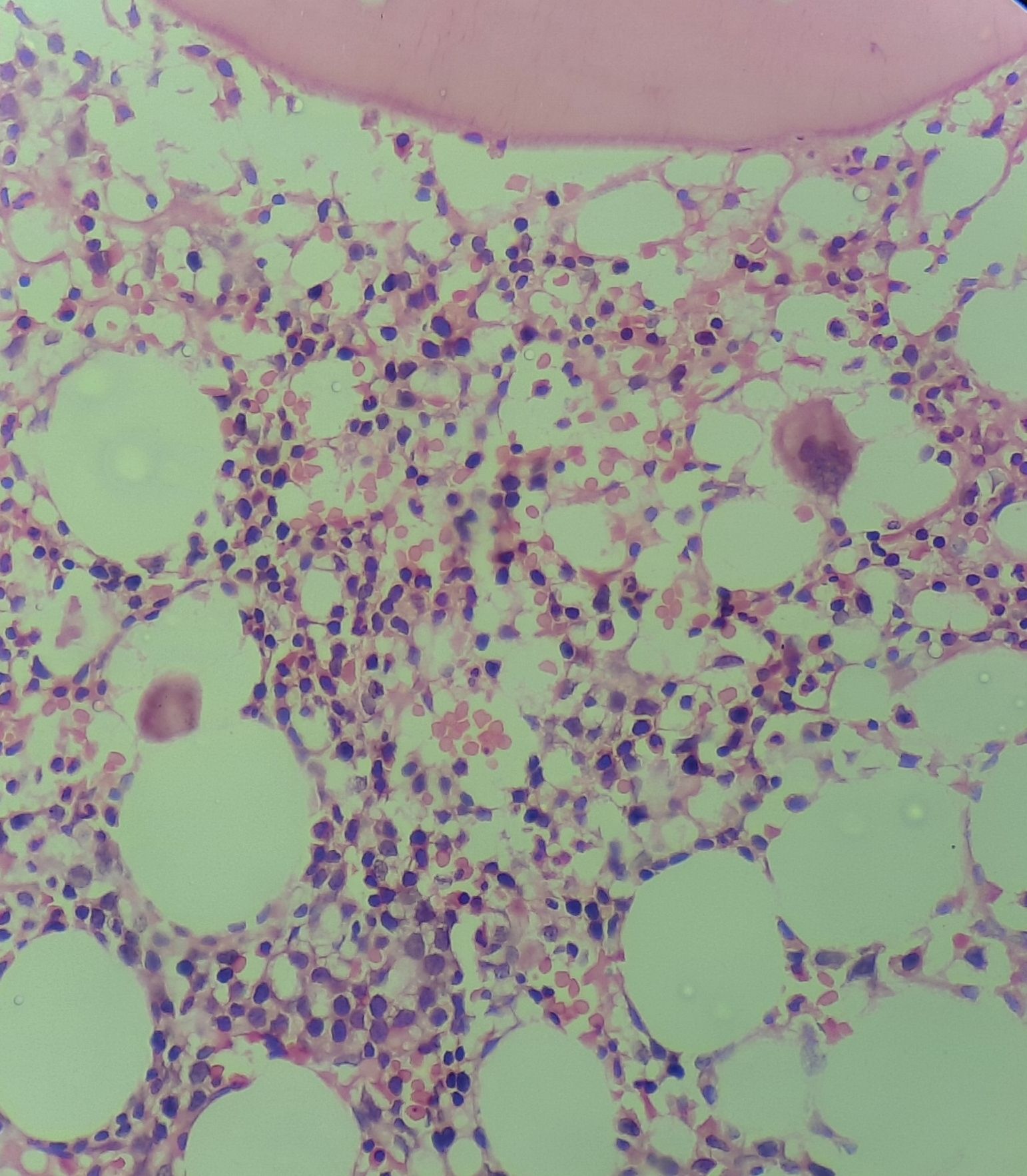
Figure 8.21.5- Autoimmune hemolytic anemia- Bone marrow biopsy
Recent advances:
Sutimlimab in patients with cold agglutinin disease: results of the randomized placebo-controlled phase 3 CADENZA trial
Sutimlimab, a first-in-class humanized immunoglobulin G4 (IgG4) monoclonal antibody that selectively inhibits the classical complement pathway at C1s. CADENZA was a 26-week randomized, placebo-controlled phase 3 study to assess safety and efficacy of sutimlimab in patients with CAD. Sutimlimab, but not placebo, significantly increased mean hemoglobin and FACIT-Fatigue scores at treatment assessment timepoint.
https://doi.org/10.1182/blood.2021014955
Maintenance rituximab following induction in autoimmune cytopenias
This study evaluated the effectiveness of rituximab maintenance in patients with immune thrombocytopenia (ITP), autoimmune hemolytic anemia, and Evans syndrome who had previously responded to rituximab induction but later relapsed. Sixteen patients received rituximab maintenance with a regimen of a single 375 mg/m2 dose administered at 4-month intervals, for a maximum of 6 doses. Of the patients, 15 out of 16 achieved complete response (CR), and 8 of those CR patients remained in remission. The median response duration was 43 months, and the estimated 5-year relapse-free rate was over 50%.
https://doi.org/10.1111/bjh.18814
Safety, tolerability, and activity of the active C1s antibody riliprubart in cold agglutinin disease
In a Phase 1b study, riliprubart, a second-generation classical complement inhibitor, was assessed for safety and efficacy in adult patients with cold agglutinin disease (CAD), a rare autoimmune hemolytic anemia. Twelve patients received a single IV dose of riliprubart (30 mg/kg or 15 mg/kg) and were followed for 15 weeks. Riliprubart demonstrated good tolerability with no serious adverse events reported, and improvements in hemoglobin and bilirubin levels were observed rapidly and sustained throughout the study period. These improvements were closely linked to sustained reduction in hemolytic complement activity and increased C4 levels, indicating effective control of hemolysis and anemia in CAD patients.
https://doi.org/10.1182/blood.2023022153
Primary autoimmune haemolytic anaemia is associated with increased risk of ischaemic stroke
In a binational study using data from nationwide registers in Denmark and France, researchers investigated the risk of ischaemic stroke in patients with primary autoimmune haemolytic anaemia (AIHA). The AIHA cohort comprised 5994 patients, and 81,525 age- and sex-matched comparators were included. The analysis revealed a higher risk of ischaemic stroke in the AIHA cohort compared to the general population, particularly within the first year after AIHA diagnosis, with a cause-specific hazard ratio of 2.29. However, this elevated risk decreased in subsequent years following diagnosis.
https://doi.org/10.1111/bjh.19242
Orelabrutinib in refractory/relapsed autoimmune haemolytic anaemia/Evans syndrome
Orelabrutinib is an orally administered, potent, irreversible and highly selective BTK-inhibitor. In a clinical trial investigating orelabrutinib treatment for refractory/relapsed autoimmune haemolytic anaemia (AIHA) and Evans syndrome, preliminary results showed promising responses in nine out of 12 enrolled patients. Three cases who completed the treatment and were followed up for 6 months achieved either complete or partial remission. Orelabrutinib demonstrates potential as a new second-line treatment option for refractory/relapsed AIHA and Evans syndrome, addressing the current lack of effective therapies for these conditions.
https://doi.org/10.1111/bjh.19146
Parsaclisib for the treatment of primary autoimmune hemolytic anemia
A phase 2 study of parsaclisib (selective phosphoinositide 3-kinase δ inhibitor) in 25 patients with warm AIHA, cold agglutinin disease, or mixed-type AIHA showed that 64% of patients achieved a partial or complete hemoglobin response. Responses were seen as early as week 1 and sustained during the extension period. The treatment was well tolerated, with the most common adverse events being diarrhea and pyrexia. Six patients discontinued due to adverse events, but no dose reductions were required.
https://doi.org/10.1002/ajh.27493
An Initiative of
Veenadhare Edutech Private Limited
1299, 2nd Floor, Shanta Nivas,
Beside Hotel Swan Inn, Off J.M.Road, Shivajinagar
Pune - 411005
Maharashtra – India

howitreat.in
CIN: U85190PN2022PTC210569
Email: admin@howitreat.in
Disclaimer: Information provided on this website is only for medical education purposes and not intended as medical advice. Although authors have made every effort to provide up-to-date information, the recommendations should not be considered standard of care. Responsibility for patient care resides with the doctors on the basis of their professional license, experience, and knowledge of the individual patient. For full prescribing information, including indications, contraindications, warnings, precautions, and adverse effects, please refer to the approved product label. Neither the authors nor publisher shall be liable or responsible for any loss or adverse effects allegedly arising from any information or suggestion on this website. This website is written for use of healthcare professionals only; hence person other than healthcare workers is advised to refrain from reading the content of this website.