
howitreat.in
A user-friendly, frequently updated reference guide that aligns with international guidelines and protocols.
Chronic Lymphocytic Leukemia/ Small Lymphocytic Lymphoma
Introduction:
- It is a neoplasm of monomorphic small, round B-lymphocytes in the peripheral blood, bone marrow and lymph nodes, admixed with prolymphocytes and para immunoblasts, usually expressing CD5 and CD23.
Epidemiology:
- It comprises of 6-7% of all NHL and 25% of all leukemias
- > 50years commonly affected (Median age 72years)
- Male to female ratio is 2:1
- Incidence- 4.2/1lac population per year
- Common in fair skinned populations
- Incidence is low but prevalence is high due to indolent course of disease
Etiology: Not known. Suspected agents include:
- Antigens promoting division of precursor cells
- Environmental agents associated with farming
- Increased exposure to electromagnetic field
- Ionizing radiations
- Familial- Risk in first degree relative is 2-7 times higher
Pathogenesis:
- Activation of BCR pathway through
- PI3k delta
- Spleen tyrosine kinase
- Bruton tyrosine kinase
- Activation of BCR pathway promotes B cell development, survival and proliferation
- Defective apoptosis due to over expression of anti-apoptotic proteins including bcl-2, mcl-1, bak and X linked inactivator of apoptotic protein (XIAP)
- Decreased expression of pro-apoptotic protein- bax
- Autocrine and paracrine networks involving
- B Cell activation factor
- A proliferation inducing ligand
- VEGF
- IL-4
- CD 40
- Role of micro-environment- Stromal cells provide survival signals- Cytokines, chemokines, CD40, B cell activationg factor of TNF family (BAFF), integrins and components of extra cellular matrix.
Clinical Features:
- Most of the patients are asymptomatic
- Anemia (Easy fatigability) in late stages. Causes of anemia include
- Marrow infiltration
- Myelosuppressive effects of chemotherapy
- Inhibitory cytokines
- Autoimmune hemolytic anemia
- Hypersplenism
- Poor nutritional status
- Pure red cell aplasia
- Constitutional symptoms- Loss of weight, anorexia, profound fatigue, drenching night sweats and fever
- Generalized lymphadenopathy
- Firm, discrete, painless.
- Present in 80% of cases
- May cause obstructive symptoms
- Splenomegaly- Seen in 50% of patients
- Hepatomegaly
- Rarely
- Auto immune hemolytic anemia
- Autoimmune thrombocytopenia
- Other autoimmune diseases
- Hemorrhagic manifestations
- Skin infiltration – circumscribed, raised, brownish nodule
- Other extranodal site involvement- Proptosis, involvement of prostate, gonads, percardium, lungs, GIT
- CNS manifestation due to infiltration- Headache, meningitis, cranial nerve palsy, coma
- Mediastinal syndrome
- Enlargement of tonsils
- Repeated infections due to impaired immunoglobulin production, T cell dysfunction, tumor cell elaboration of immunosuppressive cytokines (TGF-beta, CD27), reduced complement levels and neutropenia
- Exaggerated response to insect bites- especially mosquitoes
- Chronic rhinitis- Due to nasal involvement by CLL cells
Investigations:
- Hemogram
- Normocyticnormochromic anemia
- Leukocyte count is markedly increased (>2 lac / cmm)
- Marked lymphocytosis - > 90% are mature lymphocytes. They are small, monotonous cells having round nuclei; block type chromatin clumping and scanty blue cytoplasm. Some contain intracytoplasmic globules or crystalline rod shaped inclusions.
- Smear cells / Smudge cells / Basket cells – they are distorted cells which are formed during smear preparation.
- Prolymphocytes are < 2%.
- Neutropenia.
- Platelets – Reduced counts
- Bone Marrow Aspiration
- Cellularity is increased
- Mature lymphocyte count in bone marrow is > 40%
- Reduced myeloid as well as erythroid precursors.
- Trephine biopsy
- Heavy replacement of fat spaces and hematopoietic cells by mature lymphocytes
- Tumor cell infiltration is paratrabecular nodular / interstitial / diffuse / combination of these.
- Diffuse pattern with no residual fat indicates advanced CLL (Stage C of Binet)
- Proliferation center- Small cluster of prolymphocyte appearing cells (these cells express high levels of CD 20 and other B cell surface antigens)
- The value of bone marrow trephine biopsies in CLL
- Prognostic feature: Diffusely packed BM has poor prognosis
- Clarify the nature of cytopenias
- IHC can be done on biopsy sample if differential diagnosis includes other low grade NHL
- Lymph Node biopsy
- Effacement of architecture
- Pseudo follicular pattern of regularly distributed pale areas containing large cells in a dark background of small cells.
- Predominant cell is small lymphocyte, which is, slightly larger than a normal lymphocyte.
- Round nucleuswith clumped chromatin. Low mitotic activity.Cytoplasm is scanty and clear.
- Immunophenotyping (IHC/ Flow cytometry)
- Positive: CD5, CD23, CD19, CD20, CD22, CD79a, CD43, CD200, CD11C, IgM, BCL2, CD 45, BOB 1, OCT 2, PAX 5, LEF1
- Coexpression of CD 5 and CD 23 is typical of CLL
- Weak expression of Surface immunoglobulin IgM- kappa/lambda
- Negative for – CD10, Cyclin D1, FMC7, CD25, CD79b, CD 103, CD30, CD138, BCL1, BCL6 , SOX11
- ZAP (Zeta associated protein) 70 and CD38 expression indicates presence of unmutated IgVH gene and aggressive nature of disease and shorter survival. CD49d is also associated with poor prognosis.
- Cytogenetics/FISH
- Not required in patients with low Rai score who are not being treated.
- Before treatment cytogenetics should be done/ repeated as new mutations can appear
- Conventional metaphase cytogenetics is difficult in CLL as a result of very low proliferative activity of leukemic cells.
- Cytokine/CpGoligonucleotide stimulation may be utilized to promote metaphase
- Poor prognosis with- del 13q (miR-16, ARTLS1 and miR-15a gene), del 11q (ATM gene), del 17p (TP53gene) and complex karyotypes (≥3or ≥5 aberrations)
- FISH for t(11:14) must be done in all cases to rule out mantle cell lymphoma
- FISH is better option to detect above abnormalities
- Mutation status of Antigen receptor genes by sequencing
- Rearrangement of Ig heavy and light chain genes (Ig VH genes)- Unmutated status is associated with poor prognosis
- Sequencing of TP53 gene
- Mutation carries bad prognosis
- Repeat testing at relapse is recommended
- BTK and PLCG2 mutation: Should be done in case of relapsed CLL cases, who were on BTK inhibitors.
- BCL2 mutation: Should be done in case of relapsed CLL cases, who were on venetoclax.
- Serum immunoglobin levels - Hypogammaglobulinemia
- Computed tomography- For staging the disease
- Routine biochemical tests: RFT, LFT, LDH, Uric acid, Beta 2 microglobulin
- DCT, ICT- Positive in 20% patients, but AIHA is seen in 8% patients
- S. Protein electrophoresis: 5% of patients have monoclonal immunoglobulin paraprotein
- Sequencing of TP53 gene- To be done if treatment is planned
- Beta 2 microglobulin levels- Increased levels are associated with poor prognosis
- Serology- HIV, HBsAg, HCV
Criteria for Diagnosis:
For CLL
- Essential:
- Classic morphology of CLL cells and absolute B-cell count >5000/cmm
- Flow cytometry (on peripheral blood and/or bone marrow aspirate samples) showing expression of CD19, CD5, CD20, CD23 (variable) & weakly expressed monotypic light chain.
Or
- Histopathology/immunohistochemistry (on bone marrow cell clot/core biopsy, and biopsies of lymph node or other tissue samples) demonstrating CD20+/weak+, CD5+/weak+, CD23 (variable) expression and no expression of cyclin D1- (weak positivity in subset of cells in proliferative centers is allowed)
- Desirable:
- Flow cytometry: Positive for CD200, ROR1, CD43 with absence of FMC7, CD79b (can be weaklypo sitive), CD10, CD81 expression.
- Histopathology/immunohistochemistry: Positive for CD23, LEF1, CD43, MUM1 (proliferation centres) with absence of CD10, SOX11 expression.
For SLL (International workshop on CLL)
- Lymphadenopathy (>15mm)
- Typical immunophenotype
- No cytopenia due to bone marrow infiltration
- Absolute lymphocyte count- <5000/cmm
Staging:
Rai System of Staging
Stage | Features | Median Survival |
0 | Lymphocytosis alone | >10 years |
I | 0 + Lymphadenopathy | > 8 years |
II | I + Hepato-splenomegaly | < 7 years |
III | II + Anemia (Hb< 11 g/dL) | 2 – 5 years |
IV | III + Thrombocytopenia (Platelet count < 100 x 109 /L) | < 2 years |
Binet classification:
Stage A- Hb> 10 g/dL and Platelet count > 100 x 109 /L and <3 lymphoid areas involved
Stage B- Hb> 10 g/dL and Platelet count > 100 x 109 /L and >3 lymphoid areas involved
Stage C- Hb< 10 g/dL or Platelet count < 100 x 109 /L
Prognosis:
(Evaluation of prognostic markers is not recommended in whom there is no indication of treatment. Identification of TP53 abnormality is not an indication for treatment)
- Poor Prognostic Markers
- Advanced Rai stage
- TP53 abnormalities (Chr. 17p21)
- Unmutated immunoglobulin heavy chain variable region (IGHV) gene
- Advanced age
- Poor performance status
- Associated comorbidities
- Initial lymphocytosis>50,000/cmm
- Diffuse bone marrow involvement
- Presence of proliferative centers in bone marrow biopsy
- Rapid lymphocyte doubling time < 12 months
- High β2 microglobulin level
- High thymidinekinase, metalloprotease 9, IL8, IL6, SCD44, SUCAM1, SCD27
- Trisomy 12 with atypical morphology
- Complex karyotype
- CD-38 positivity in >30% lymphocytes
- ZAP 70 and CD49d positivity
- Increased serum levels of sCD23, TNF alpha, LDH
- Mutations in NOTCH 1, SF3B1, BIRC3 genes, genes of lipoprotein lipase, dysintegrin, metalloprotease 29,
- 11q 22-23 deletion with extensive lymphadenopathy
- Increased serum levels of CD23, TNFɑ &thymidine kinase
- Accelerated CLL/SLL: Histologically aggressive CLL/SLL and Prolymphocytic progression (See below)
- Better Prognosis
- Abnormalities of 13q14
- Mutations in Ig genes variable regions
- IgD+/ IgM- phenotype
- Overall
- CLL has an indolent course
- 5 year survival is 51%
- Overall median survival is 7 years
- Disease is not curable by conventional therapies
- CLL International prognostic index:
Variable | Adverse factor | Score |
Age | >65 years | 1 |
Clinical stage | Binet B/C or Rai I-IV | 1 |
17p deletion/ TP53 mutation | Deleted/ Mutated | 4 |
IGHV mutation status | Unmutated | 2 |
Beta 2 microglobulin level | >3.5mg/L | 2 |
Score | Risk | OS at 5 years |
0-1 | Low | 93.2% |
2-3 | Intermediate | 79.3% |
4-6 | High | 63.3% |
7-10 | Very high | 23.3% |
Indications for Treatment:
(These criteria apply to both previously untreated and relapsed patients)
- Eligibility for clinical trial (treatment based on newer prognostic markers)
- Symptomatic patents- Fever, weight loss (>10% in last 6 months)
- Fatigue with PS 2 or worse
- Obstructive or advanced lymphadenopathy
- Massive splenomegaly (>6cm below costal line)
- Stage III or IV disease i.e. Hemoglobin <10gm/dL or Platelet count <1lac/cmm(Which is due to marrow infiltration and not due to other causes)
- Lymphocyte doubling time <6months (Baseline ALC must be >30,000/cmm)
- Complications- Recurrent infections, threatened end organ damage, transformation to DLBCL
- AIHA or ITP which are unresponsive to steroids
- Paraneoplastic symptoms which are not responding to traditional treatment. Ex: Insect bite hypersensitivity, vasculitis, myositis
Pretreatment Work-up (Do if indication for treatment present):
- History
- B-Symptoms
- Examination
- LN:
- Spleen:
- WHO P. S.
- BSA
- IHC/Flow cytometry
- CD 38
- ZAP 70
- BMA and Bx
- Hemoglobin
- TLC, DLC
- Platelet count
- Peripheral smear
- Reticulucyte count
- DCT
- LFT: Bili- T/D SGPT: SGOT: Albumin: Globulin:
- Creatinine
- Electrolytes: Na: K: Ca: Mg: PO4:
- Uric acid
- LDH
- β2 microglobulin
- HIV
- HBsAg
- HCV
- UPT
- Quantitative Ig
- CpG stimulated Cytogenetics
- PET-CT (If maximum standardized uptake value of >10 in any area, a diagnostic biopsy should be taken to rule out Ritcher transformation)
- FISH
- Trisomy 12
- del(11q)
- del(13q)
- del(17p)
- t (11:14) – To rule out mantle cell lymphoma
- TP53 Sequencing
- IGHV Mutation
- BTK, PLCG2 and BCL2 mutation analysis
- Prognostic score
- ECHO (If anthracyclines planned) LVEF- %
- Chemotherapy consent after informing about disease, prognosis, cost of therapy, side effects, hygiene, food and contraception
- Fertility preservation
- PICC line insertion and Chest X ray after line insertion
- Tumor board meeting and decision
- Attach supportive care drug sheet
- Inform primary care physician
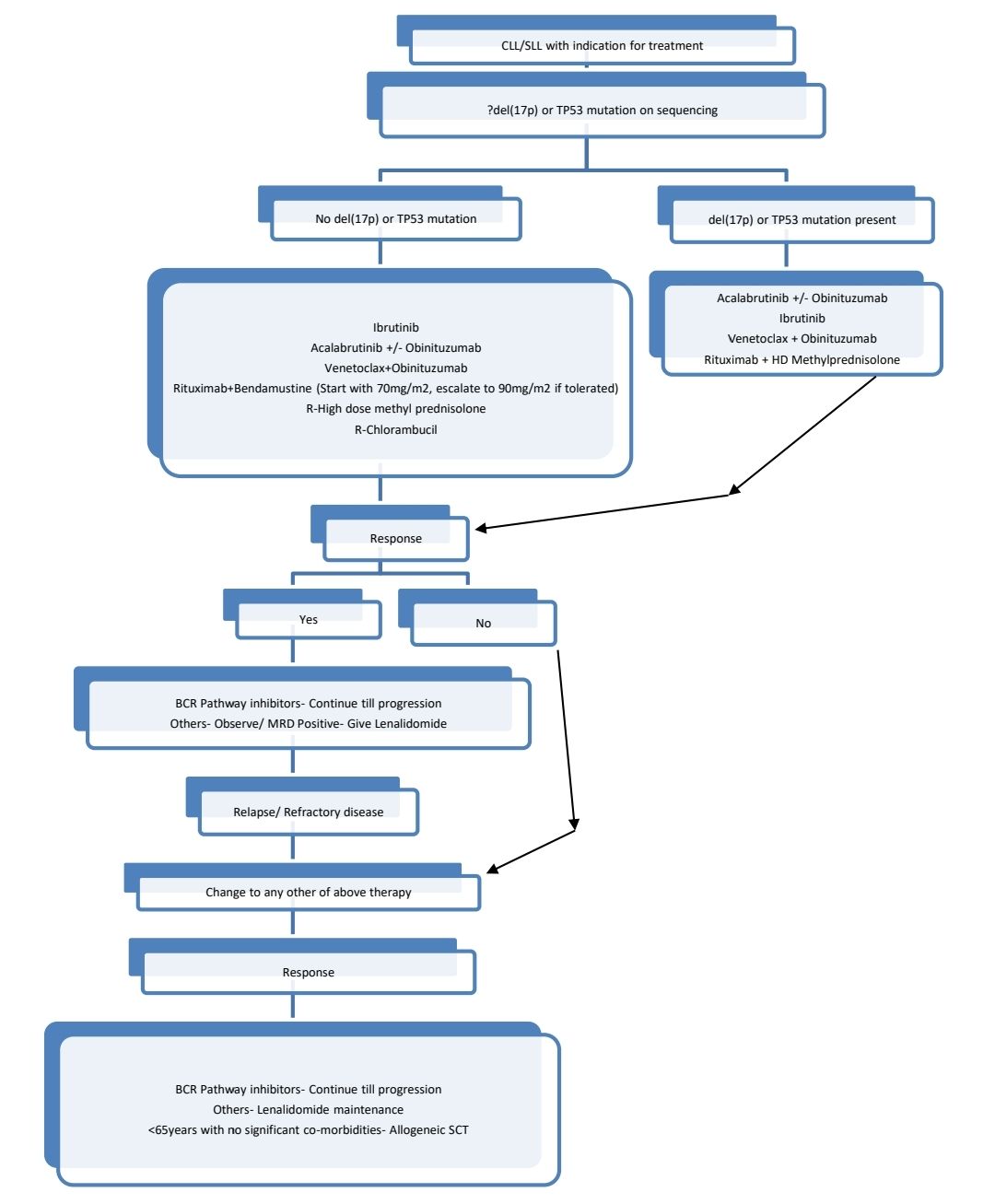
Treatment Plan:
SLL- Localised disease-Loco-regional RT (24-30 Gy)- then oberve
Rest all SLL and all CLL:
Response Criteria:
- To be assessed 2 months after completion of treatment
- Criteria for complete response: (All criteria are met with no symptoms)
- Group A (Based on tumor load)
- Lymphadenopathy- None >1.5cm
- No hepatomegaly
- No splenomegaly
- BM- Normocellular with <30% lymphocytes, no B lymphoid nodules (Hypocellular marrow indicates complete response but incomplete marrow recovery)
- Absolute lymphocyte count <4000/cmm
- Group B (Based on marrow function)
- Platelet count (unsupported)- >1lac/cmm
- Hemoglobin (unsupported)- >11gm/dL
- ANC (unsupported))- >1500/cmm
About Each Modality of Treatment:
- Ibrutinib
- Orally active irreversible inhibitor of BTK
- In patients with CLL- Dose- 420mg- OD
- After starting, there can be transient increase in lymphocyte count (may persist for several weeks)
- Avoid co-administration of CYP3A inhibitors
- Side effects- Bleeding episodes, diarrhea, skin rashes
- Chemotherapies
- Refer to NHL section
- Allo HSCT in CLL
- Indication-
- No response/ Relapse after BCR pathway inhibitor
- Eligible patients with Ritcher transformation
- With related HSCT- CR rate- 55%
- Indication-
Supportive Care:
- Treatment of AIHA/ITP
Prednisolone- 1mg/kg/day (for 2-6 weeks then taper over 3 months)
↓
No response
↓
Ciclosporine- 2.5mg/kg- BD (Target serum level- 100-150)
↓
No response
↓
Rituximab- 375mg/m2- IV- Weekly for 4 weeks/ Eltrombopag in ITP
↓
No response
↓
Treatment of CLL
- Repeated infections with hypogammaglobulinemia - IVIg- 400mg/kg- every 3-4 weeks
- Give prophylactic Acyclovir, Septran during treatment and up to 4 months after completion of therapy
- Annual influenza vaccine, Pneumococcal vaccine once in 5 years
Other Treatment Options:
- Novel agents under investigation
- BCR pathway inhibitors:
- SYK Inhibitors- Fostamitinib, GS-9973
- BTK Inhibitors- CC-292, ONO- 4059, ACP-196
- PI3K Inhibitors- IPI-145, AMG-319
- BCR pathway inhibitors:
- Flavopiridol
- Lenalidomide
- Ofatumumab
- Active immune therapy using cellular vaccines/ modified leukemia cells
- Gene therapy
- CAR-T cell therapy
- BCL2 antisense molecule- Navitoclax
- PI3 Kinase inhibitor- GS 1101
- Irreversible inhibitor of Btk- PCI 32765
Monitoring After Treatment/ Follow-up:
- For patients with no indication for treatment- Follow up once in 6 months with CBC
- For patients who have completed treatment- Once in 3 months with CBC
Transformations of CLL/SLL:
- Accelerated CLL/SLL
- Histologically aggressive CLL/SLL
- Prolymphocytic progression
- Richter transformation
Histologically aggressive CLL/SLL
- Have very large, prominent or confluent proliferation centers spanning a diameter of a visual field using 20X objective lens and 10X ocular lens or high proliferation index (Ki67- >40%)
Prolymphocytic progression
- Number of prolymphocytes is increased to >15%
- Includes those cases which were previously called prolymphocytic leukemia (Cases with >55% prolymphocytes)
- Their increased number correlates with aggressive disease course, p53 mutation and trisomy 12.
- Present with Fatigue, weakness, weight loss, scquired bleeding tendency and splenomegaly (usually massive) without lymphadenopathy.
- Hemogram
- Total leukocyte count- Usually >1lac/cmm
- Prolymphocytes measure double the size of small lymphocyte. Nucleus is round with condensed nuclear chromatin and prominent central nucleolus. Cytoplasm is scanty & basophilic.
- Lymph node biopsy: Diffuse nodular infiltration
- Immunophenotyping
- Strongly positive- surface IgM, IgD, CD19, CD20, CD22,CD79a&b, FMC7
- Negative- CD5, CD23
- Molecular studies
- Clonal rearrangement of immunoglobulin gene
- Break points involving 14q32-t(11:14)
- Abnormalities of TP53 gene
- S. Immunoglobulin levels- Decreased
- FISH for t(11;14)- Done to rule out mantle cell lymphoma
- Rapid progression and poor response to treatment
- Denovo B-PLL (CD5 -ve) has better prognosis compared to B-PLL arising from B-CLL (CD5+ve)
- Indications for Treatment: Same as those of CLL
- Disease related symptoms
- Symptomatic splenomegaly
- Progressive marrow failure
- Blood prolymphocyte count- >2lac/cmm
- Most of the patients need treatment, as most of the patients present in advanced stage of disease and also disease has rapid progression.
- Treatment:
- Chemotherapy (Responses are short lasting)
- R-CHOP- Response rate 50%
- R- FC
- Cladribine (Same as HCL)
- CVP- Response rate <20%
- Splenectomy- Amelioration of symptoms for short time
- Splenic irradiation- For patients considered poor candidates for chemotherapy/ splenectomy
- Allo SCT is the only way to cure. It can be tried in selected fit patients.
- Chemotherapy (Responses are short lasting)
Ritcher transformation
- It is transformation of an isolated lymph node affected by CLL into an aggressive lymphoma.
- Transformations include
- Diffuse large B cell lymphoma- 90% (Usually non-GCB type)
- Hodgkin’s lymphoma- 10%
- Rarely- Histiocytic/ dendritic cell sarcoma/ other lymphoma.
- Most commonly it occurs due to an additional p53 mutation.
- Other genetic changes include del (11q), Overexpression of c-myc, deletion of Rb1
- It presents with fever, increasing lymphadenopathy at a perticular site, weight loss, abdominal pain and sudden rise in LDH levels. Rarely there can be hepatomegaly and splenomegaly.
- It is seen in 3-5% cases of CLL
- Immunosuppression with fludarabine triggers this transformation by causing CD4 + lymphopenia
- There is evidence of involvement of EBV in pathogenesis (LMP is demonstrated by IHC and EBER by FISH).
- Diagnosis requires lymph node biopsy
- Prognosis- Poor
- Treatment- Same as DLBCL
Related Disorders:
- Familial CLL
- Relative risk of developing CLL in patient’s relative is 3-5 times higher than normal population.
- Anticipation is usually noted – early onset and more severe disease in successive generations.
- No clear cut candidate gene has yet emerged.
- Atypical B- CLL
- Larger lymphocytes with abundant cytoplasm
- Prolymphocyte like/ cleaved cells
- CoexpressIgM and IgD
- Monoclonal B Lymphocytosis (MBL)
- Clonal B cell count is <5000/cmm with no disease related cytopenia, B symptoms, lymphadenopathy or splenomegaly
- 2 subtypes:
- CLL/SLL phenotype
- Non CLL/SLL phenotype
- Seen in younger individuals and patients after chemotherapy
- 0.5- 2% individuals with CLL/SLL phenotype MBL progress to CLL/SLL every year
- Treatment: Observe
Figures:
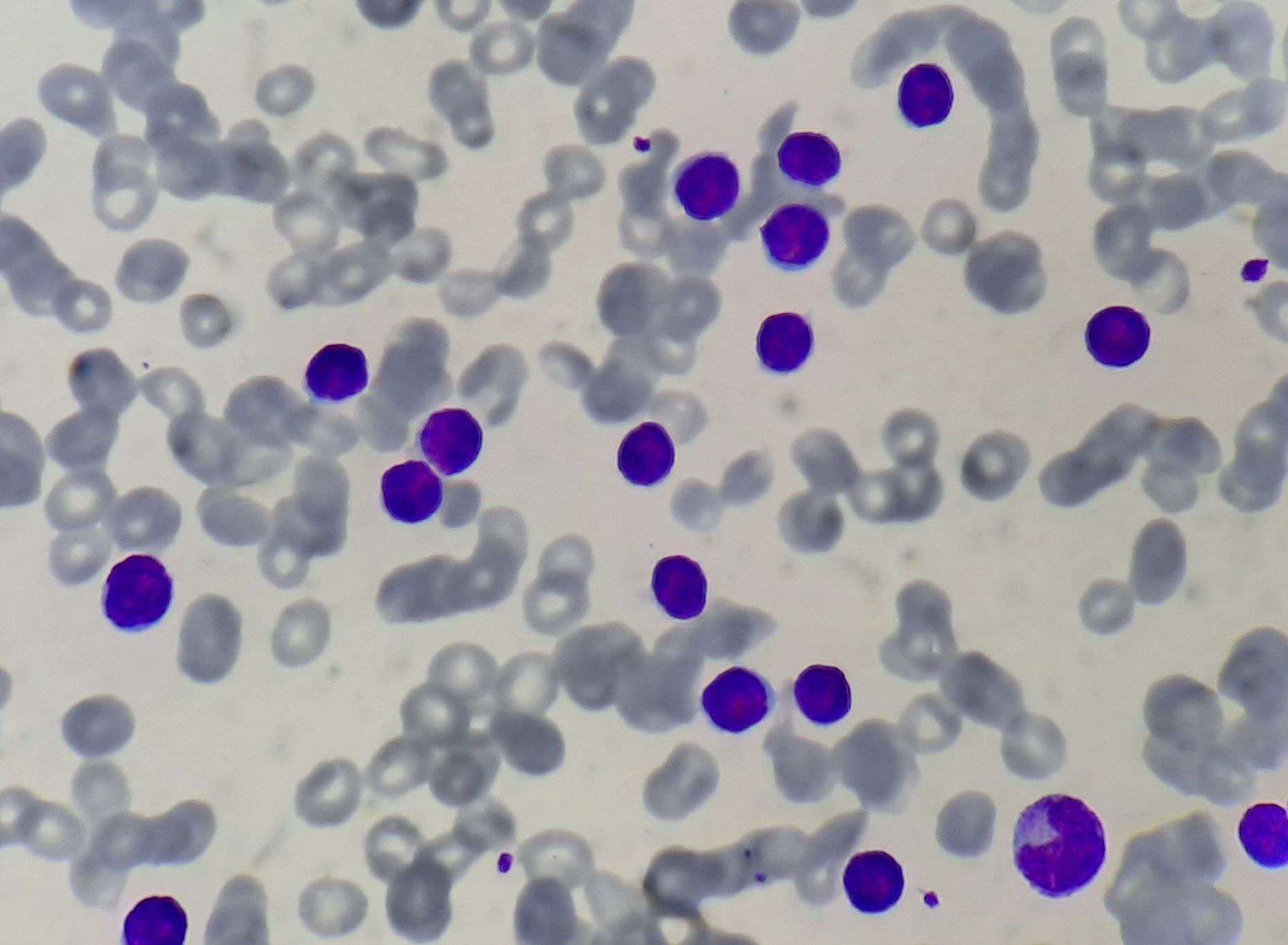
Figure- 6.2.1- Chronic lymphocytic leukemia- Peripheral smear
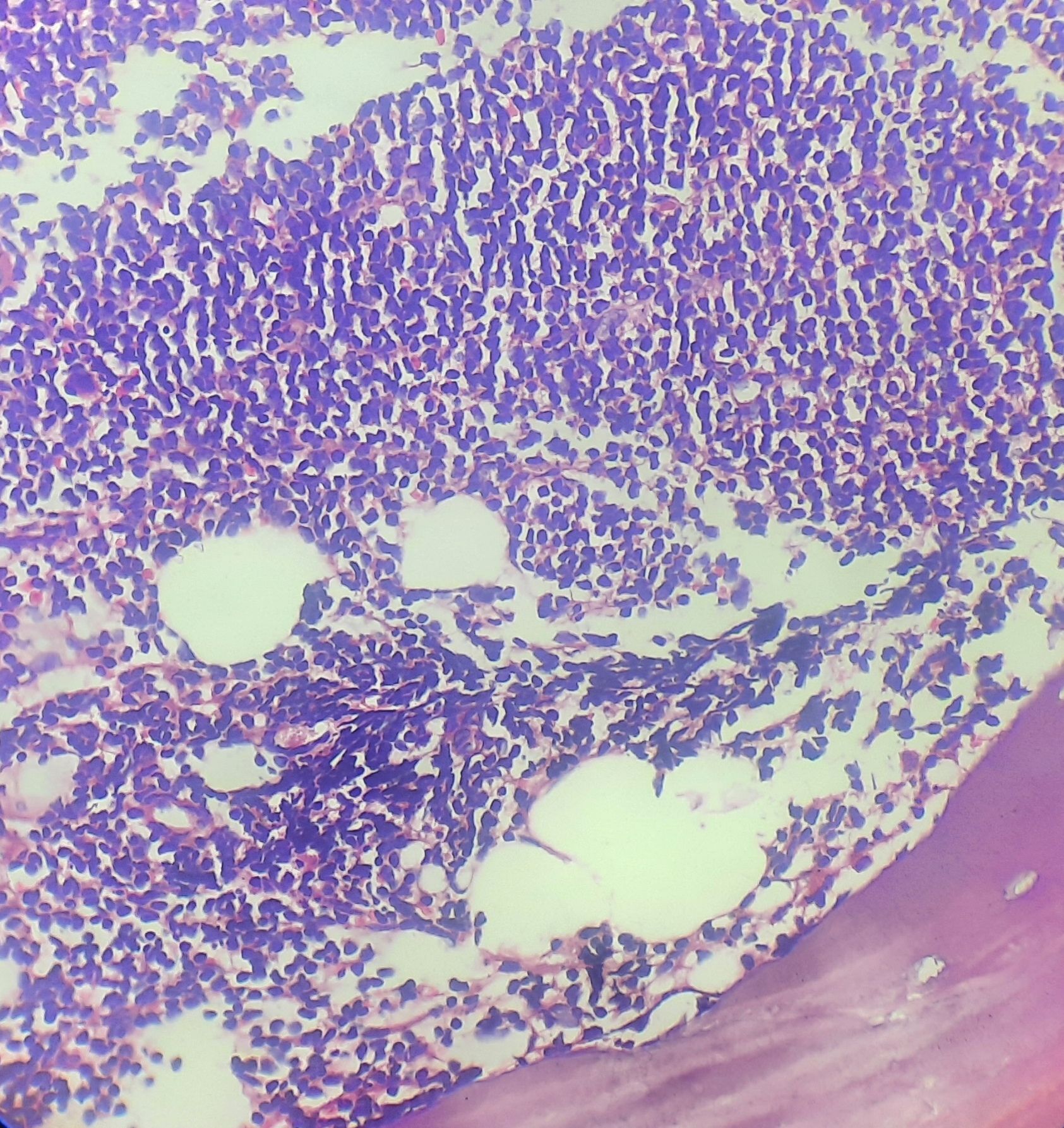
Figure- 6.2.2- Chronic lymphocytic leukemia- Bone marrow biopsy
Recent advances:
Fixed-duration ibrutinib plus venetoclax for first-line treatment of CLL
CAPTIVATE is an international phase 2 study in patients aged ≤70 years with previously untreated chronic lymphocytic leukemia. Patients received 3 cycles of ibrutinib lead-in then 12 cycles of ibrutinib plus venetoclax (oral ibrutinib [420 mg/d]; oral venetoclax [5-week ramp-up to 400 mg/d]). Of the 159 patients enrolled and treated, 136 were without del(17p). The primary endpoint was met, with a CR rate of 56% in patients without del(17p), significantly higher than the prespecified 37% minimum rate . In the all-treated population, CR rate was 55%. First-line ibrutinib plus venetoclax represents the first all-oral, once-daily, chemotherapy-free regimen for patients with CLL. Fixed duration ibrutinib plus venetoclax achieved deep, durable responses and promising PFS, including in patients with high-risk features.
https://doi.org/10.1182/blood.2021014488
Zanubrutinib or Ibrutinib in Relapsed or Refractory Chronic Lymphocytic Leukemia
In present study patients with relapsed or refractory CLL or SLL were randomy assigned to receive zanubrutinib or ibrutinib until the occurrence of disease progression or unacceptable toxic effects. At a median follow-up of 29.6 months, zanubrutinib was found to be superior to ibrutinib with respect to progression-free survival among 652 patients. The safety profile of zanubrutinib was better than that of ibrutinib.
https://doi.org/10.1056/NEJMoa2211582
Zanubrutinibin Relapsed/Refractory Chronic Lymphocytic Leukemia
Compared with ibrutinib, zanubrutinib, a next-generation BTKi, which provides improved BTK occupancy across disease-relevant tissues with greater kinase selectivity. In present randomized phase 3 study, zanubrutinib was compared head-to-head with ibrutinib as treatment for R/R CLL/SLL. With a median follow-up of 29.6 mo zanubrutinib PFS, assessed by independent review committee, was superior to ibrutinib.
https://doi.org/10.1182/blood-2022-171538
First-Line Venetoclax Combinations in Chronic Lymphocytic Leukemia
In this study 926 patients were randomly assigned, in a 1:1:1:1 ratio, to receive six cycles of chemoimmunotherapy (fludarabine–cyclophosphamide–rituximab or bendamustine–rituximab) or 12 cycles of venetoclax–rituximab, venetoclax–obinutuzumab, or venetoclax–obinutuzumab–ibrutinib. The primary end points were undetectable minimal residual disease (sensitivity, <10−4 [i.e., <1 CLL cell in 10,000 leukocytes]) as assessed by flow cytometry in peripheral blood at month 15 and progression-free survival. At month 15, the percentage of patients with undetectable minimal residual disease was significantly higher in the venetoclax–obinutuzumab group. Three-year progression-free survival was 90.5% in the venetoclax–obinutuzumab–ibrutinib group and 75.5% in the chemoimmunotherapy group.
https://doi.org/10.1056/NEJMoa2213093
Ibrutinib in combination with rituximab is highly effective in treatment of chronic lymphocytic leukemia patients with steroid refractory and relapsed autoimmune cytopenias
This multicenter study investigated the use of ibrutinib and rituximab in patients with relapsed or refractory autoimmune hemolytic anemia (AIHA) and pure red cell aplasia (PRCA) associated with chronic lymphocytic leukemia (CLL). Among the 50 recruited patients, 74% achieved a complete response and 21.7% achieved a partial response after induction treatment. The median time to hemoglobin normalization was 85 days. In terms of CLL response, 19% achieved a complete response, 4% had stabilization, and 78% had a partial response. The combination of ibrutinib and rituximab demonstrated efficacy as a second-line treatment option for patients with relapsed or refractory AIHA/PRCA and underlying CLL.
https://doi.org/10.1038/s41375-023-01891-3
Idelalisib plus rituximab versus ibrutinib in the treatment of relapsed/refractory chronic lymphocytic leukaemia
This study analyzed 171 patients treated with R-idela and 244 patients treated with ibrutinib. The ibrutinib group showed significantly longer median progression-free survival (PFS) and overall survival (OS) compared to the R-idela group. Furthermore, ibrutinib demonstrated better tolerability, with fewer cases of treatment discontinuation due to toxicity or CLL progression.
https://doi.org/10.1111/bjh.18736
Obinutuzumab, ibrutinib, and venetoclax for untreated CLL with del(17p)/TP53mut
The CLL2-GIVe trial examined the triple combination of obinutuzumab, ibrutinib, and venetoclax (GIVe regimen) in 41 previously untreated patients with high-risk chronic lymphocytic leukemia (CLL) with del(17p) and/or TP53 mutation. After a median observation time of 38.4 months, the complete remission rate at cycle 15 was 58.5%. The 36-month progression-free survival was 79.9%, and the 36-month overall survival was 92.6%. Adverse events included neutropenia (48.8%, grade ≥3) and infections (19.5%, grade ≥3), with cardiovascular toxicity (atrial fibrillation and hypertension) occurring at rates of 2.4% and 4.9%, respectively. Adverse events were most common during induction and decreased over time. The regimen appears promising as a first-line treatment with a manageable safety profile for high-risk CLL patients.
https://doi.org/10.1182/blood.2023020013
Sustained remissions in CLL after frontline FCR treatment with very-long-term follow-up
Chemoimmunotherapy with fludarabine, cyclophosphamide, and rituximab (FCR) has shown durable remissions, particularly in patients with mutated immunoglobulin heavy chain variable gene (IGHV-M). In a study initiated in 1999 with a median follow-up of 19.0 years, patients with IGHV-M had a median progression-free survival (PFS) of 14.6 years compared to 4.2 years for those with unmutated IGHV (IGHV-UM). While disease progression beyond 10 years was uncommon, therapy-related myeloid neoplasms (tMNs) occurred in 6.3% of patients, emphasizing the importance of a risk-benefit assessment when considering FCR for chronic lymphocytic leukemia (CLL), weighing potential functional cure against the risk of late relapses and serious secondary malignancies.
https://doi.org/10.1182/blood.2023020158
Chronic Lymphocytic Leukemia Therapy Guided by Measurable Residual Disease
In a phase 3, multicenter, randomized trial involving patients with untreated chronic lymphocytic leukemia (CLL), the combination of ibrutinib and venetoclax was compared to fludarabine–cyclophosphamide–rituximab (FCR). The ibrutinib–venetoclax group showed improved progression-free survival compared to the FCR group, with a hazard ratio of 0.13. The risk of death was also lower in the ibrutinib–venetoclax group. MRD-directed ibrutinib–venetoclax therapy resulted in undetectable MRD in a significant percentage of patients. Although the risk of infection was similar, the ibrutinib–venetoclax group had a higher percentage of patients with cardiac serious adverse events. The study suggests that MRD-directed ibrutinib–venetoclax is a promising treatment for untreated CLL.
https://doi.org/10.1056/NEJMoa2310063
Third-generation anti-CD19 CAR T cells for relapsed/refractory chronic lymphocytic leukemia
In this phase 1/2 trial, nine heavily pretreated patients with relapsed or refractory CLL received third-generation HD-CAR-1 CD19-targeted CAR T-cell therapy. With successful in-house manufacturing, 67% of patients achieved complete remission by day 90, and 83% of these achieved undetectable MRD. After 27 months of follow-up, the 2-year progression-free survival and overall survival rates were 30% and 69%, respectively. The therapy showed promising efficacy with minimal toxicity, suggesting HD-CAR-1 as a viable option for r/r CLL.
https://doi.org/10.1038/s41375-024-02392-7
Fixed-duration pirtobrutinib plus venetoclax with or without rituximab in relapsed/refractory CLL
In a phase 1b trial, pirtobrutinib, a selective, noncovalent Bruton tyrosine kinase inhibitor, was combined with venetoclax (PV) or venetoclax and rituximab (PVR) to treat patients with relapsed or refractory chronic lymphocytic leukemia (CLL), including those previously treated with a covalent BTKi. The treatment showed high efficacy, with overall response rates of 93.3% for PV and 100% for PVR, and significant reductions in minimal residual disease, with 85.7% of PV patients and 90.0% of PVR patients achieving undetectable disease after 12 cycles. The combination therapies were well tolerated, with manageable adverse events, and demonstrated promising progression-free survival rates, particularly for PV patients at 92.9% at 18 months.
https://doi.org/10.1182/blood.2024024510
Improved efficacy and safety of zanubrutinib versus ibrutinib in patients with relapsed/refractory chronic lymphocytic leukemia
The ALPINE trial evaluated zanubrutinib versus ibrutinib in Chinese patients with relapsed/refractory (R/R) CLL/SLL. Among 90 patients, zanubrutinib showed higher overall response rates (80.9% vs. 72.1%) and improved progression-free survival (HR = 0.34) compared to ibrutinib, with a favorable safety profile. Rates of serious treatment-emergent adverse events were lower with zanubrutinib (35.6% vs. 51.2%). These findings align with global ALPINE results, supporting zanubrutinib's efficacy and tolerability.
https://doi.org/10.1007/s00277-024-05823-8
Sustained benefit of zanubrutinib vs ibrutinib in patients with R/R CLL/SLL
The ALPINE trial, with a median follow-up of 42.5 months, demonstrated that zanubrutinib was superior to ibrutinib in patients with relapsed/refractory chronic lymphocytic leukemia and small lymphocytic lymphoma, showing a progression-free survival benefit (HR, 0.68). Zanubrutinib also had a higher overall response rate (85.6% vs 75.4%) and deeper responses over time, including higher complete response rates. Although overall survival has not been reached, zanubrutinib patients experienced fewer deaths (HR, 0.77) and lower rates of cardiac events, including atrial fibrillation/flutter. Nonhematologic adverse events were comparable, but zanubrutinib showed an improved safety profile, especially regarding cardiac events.
https://doi.org/10.1182/blood.2024024667
An Initiative of
Veenadhare Edutech Private Limited
1299, 2nd Floor, Shanta Nivas,
Beside Hotel Swan Inn, Off J.M.Road, Shivajinagar
Pune - 411005
Maharashtra – India

howitreat.in
CIN: U85190PN2022PTC210569
Email: admin@howitreat.in
Disclaimer: Information provided on this website is only for medical education purposes and not intended as medical advice. Although authors have made every effort to provide up-to-date information, the recommendations should not be considered standard of care. Responsibility for patient care resides with the doctors on the basis of their professional license, experience, and knowledge of the individual patient. For full prescribing information, including indications, contraindications, warnings, precautions, and adverse effects, please refer to the approved product label. Neither the authors nor publisher shall be liable or responsible for any loss or adverse effects allegedly arising from any information or suggestion on this website. This website is written for use of healthcare professionals only; hence person other than healthcare workers is advised to refrain from reading the content of this website.