
howitreat.in
A user-friendly, frequently updated reference guide that aligns with international guidelines and protocols.
Myelodysplastic/ Myeloproliferative neoplasms (MDS/MPN)
Introduction:
- These are myeloid disorders that have both dysplastic and proliferative features at the time of initial presentation.
- They include
- Chronic myelomonocytic leukemia
- MDS/MPN with neutrophilia (Previously called atypical CML)
- MDS/MPN with SF3B1 mutation and thrombocytosis
- Myelodysplastic/myeloproliferative neoplasm, NOS
Chronic Myelomonocytic Leukemia
Introduction:
- It is a clonal disorder of bone marrow stem cell with overlapping features of myelodysplastic syndrome and myeloproliferative disorders, in which monocytosis (>1000/cmm) is a major defining feature.
Epidemiology:
- Account for 31% of total MDS
- Incidence- 4/1,00,000 per year
- Common at 65-75 years
- Male predominance
Pathogenesis:
- Various genetic mutations lead to hypersensitivity to G-CSF
- Bone marrow has inflammatory cytokine microenvironment
Clinical Features:
- Asymptomatic
- Proliferative symptoms (Leucocytosis related)
- Fatigue, weight loss, fever and night sweats
- Bone pains
- Splenomegaly and Hepatomegaly
- Maculopapular skin infiltration- Leukemia cutis
- Renal dysfunction due to high lysozyme levels
- Monocytic pleural/ pericardial effusion
- Dysplastic symptoms (Cytopenia related)
- Fatigue
- Infections
- Bleeding/ Bruising
- Immune manifestations (Seen in 30% patients)- Vasculitis, pyodermagangenosum, immune cytopenia, connective tissue disease, Rheumatoid arthritis
- Lymphadenopathy- Uncommon, but presence indicates probable transformation to more acute phase
Investigations:
- Hemogram:
- Monocytes- >10% in differential count, Absolute count >500/cmm
- Monocytes are mature with unremarkable morphology (Sometime show abnormal granulation, nuclear lobation, finely dispersed nuclear chromatin)
- Blasts &promonocytes may be seen – But their count is <20%
- Promonocytes are monocytic precursors with abundant light gray or slightly basophilic cytoplasm with a few scattered, fine iliac-colored granules, finely-distributed, stippled nuclear chromatin, variably prominent nucleoli, and delicate nuclear folding or creasing.
- Neutrophilia
- Dysgranulopoiesis – Neutrophils with hypolobulated nuclei or abnormal cytoplasmic granulation (seen especially when counts are reduced)
- Mild basophilia, sometimes eosinophilia
- Mild normocytic anemia
- Moderate thrombocytopenia – Atypical large platelets may be seen
- Bone marrow aspiration and biopsy
- Hypercellular (in > 75% of cases)
- Granulocytic proliferation- Sometimes associated with dysgranulopoiesis
- Erythroid precursors- Increased/ decreased, sometimes with megaloblastic changes
- Monocytic proliferation invariably present (Identified by alpha-naphthyl butyrate esterase)
- Megakaryocytes – Micromegakaryocytes or megakaryocytes with abnormal lobulated nuclei
- Fibrosis – Variable degree (Seen in 30% patients)
- Nodules composed of mature plasmacytoid dendritic cells- Seen in 20% cases
- Starry sky pattern with histiocytes containing apoptotic bodies may be seen in some cases.
- Serum and urinary lysozyme levels – elevated
- Cytochemistry: Monocytes are positive for Non specific esterases and lysozymes.
- Immunophenotyping
- Tumour cells are strongly positive for mature monocytic markers- CD11b, CD11c, CD14, CD33, CD45, CD64.
- Aberrant expression:
- Positive for CD2, CD15, CD56
- Negative for CD14, CD13, HLA-DR, CD64, or CD36.
- Most reliable IHC markers include CD68R and CD163.
- 3 types of monocytes can be identified by flow cytometry (Partition)
- Classical monocytes (M01)- CD14+/CD16-
- Intermediate monocytes (M02)- CD14+/ CD16+
- Non-classical monocytes (M03)- CD14-Low/ CD16+
- In CMML- M01 type monocytes are increased (Usually >90%). Their number is decreased in reactive monocytosis.
- Myelomonocytic antigens - CD33 & CD13 (sometimes CD14, CD68, CD64)
- Increased CD34 + cells is associated with early transformation to acute leukemias
- Cytogenetics
- Nothing specific
- Abnormality is seen in 20 – 40% cases which include trisomy 8,-Y, -7 / del (7q), Structural abnormalities of 12p
- t (5:12) – Results in abnormal fusion gene PDGFR. This is associated with marked eosinophilia (Now considered as separate entity)
- Molecular studies by NGS panel from peripheral blood sample (>90% have somatic gene mutations)
- Mutations in epigenetic control of transcription,such as histone modification (EZH2, ASXL1, UTX), and DNA methylation (TET2, DNMT3A, IDH1, and IDH2)
- Mutations in the spliceosome machinery (SF3B1, SRSF2, U2AF1, ZRSR2, PRPF8)
- Mutations in genes that regulate cell signaling (JAK2, KRAS, NRAS, CBL, PTPN11, and FLT3)
- Mutations in transcription factors and nucleosome assembly regulators (RUNX1, SETBP1)
- Mutations in DNA damage response genes such as TP53 and PHF6
- TET2,ASXL1, SRSF2 and RAS gene mutation are common
- Hypermethylation of p15 gene – seen in 50% cases
- Mutation of Fibroblast growth factor receptor 1 gene on chr. 9p11
Criteria for Diagnosis:
- Essential criteria
- Persistent absolute (≥ 500/cmm) and relative (≥ 10%) peripheral blood monocytosis.
- Blasts constitute < 20% of the cells in the peripheral blood and bone marrow.
- Not meeting diagnostic criteria of chronic myeloid leukemia or other myeloproliferative neoplasms.
- Not meeting diagnostic criteria of myeloid/lymphoid neoplasms with eosinophilia and defining gene rearrangements (e.g. PDGFRA, PDGFRB, FGFR1, or JAK2).
- Desirable criteria
- Dysplasia involving ≥ 1 myeloid lineages.
- Acquired clonal cytogenetic or molecular abnormality.
- Abnormal partitioning of peripheral blood monocyte subsets.
- Requirements for diagnosis
- Essential criteria must be present in all cases.
- If monocytosis is ≥ 1,000/cmm: one or more desirable criteria must be met.
- If monocytosis is <1,000/cmm: desirable criteria 1 and 2 must be met.
Types of CMML (based on percentage of blasts and promonocytes)
- CMML-1: < 5% in peripheral blood and/or < 10% in bone marrow
- CMML-2:
- 6-19% in peripheral blood and/or 10-19% in bone marrow
- Auer rods are present in blasts (Irrespective of blast percentage)
- CMML with eosinophilia
- Criteria for CMML are present + Peripheral blood Eosinophil count-> 1.5 x 109 / L
- Patients have systemic complications due to degranulation from eosinophils
FAB Classification
- Myelodysplastic CMML: WBC Count- <13,000/cmm
- Myeloproliferative CMML- >13,000/cmm
Prognosis:
- Poor prognostic factors are
- Anemia
- Thrombocytopenia
- Lymphocytosis
- Raised serum LDH
- Increased percentage of BM blasts
- Old age and poor performance score
- Median survival: 20 – 40 months
- Progression to AML occurs in 15-30% patients
- Mayo molecular model
- 1 Point each assigned to
- Absolute monocyte count- >10,000/cmm
- Circulating immature cells- myeloblasts, myelocytes, metamyelocytes and/or promyelocytes
- Hemoglobin - <10gm/dL
- Platelet count- <1,00,000/cmm
- ASXL1 frameshift or nonsense mutation
Points | Risk | Overall survival |
0 | Low | 97 months |
1 | Intermediate-1 | 59 months |
2 | Intermediate-2 | 31 months |
>2 | High | 16 months |
Other risk starification methods:
- CPSS-Mol score (Includes CMML- FAB subtype, Blasts in BM, Hemoglobin, Cytogenetics, mutations observed)
- GFM Model (Includes age, WBC count, hemoglobin, platelet count and ASXL mutation status)
Indications for treatment:
- Excess of blasts in blood or bone marrow
- Symptoms present
- Hemoglobin- <10gm/dL
- Platelet count <30,000/cmm or bleeding symptoms
- Symptomatic splenomegaly/ Other extramedullary disease- Typically cutaneous involvement or serous effusions
Pretreatment Work-up:
- History
- Examination: LN:Spleen: Skin lesions
- WHO P. S.
- BSA
- Flow cytometry
- BMA and Bx
- Haemoglobin
- TLC, DLC
- Abs Monocyte Count
- Platelet count
- Peripheral smear
- LFT: Bili- T/D SGPT: SGOT: Albumin: Globulin:
- Creatinine
- Electrolytes: Na: K: Ca:Mg: PO4:
- Uric acid
- LDH
- HIV
- HBsAg
- HCV
- UPT
- USG Abdomen- Spleen size
- Cytogenetics
- BCR-ABL1
- JAK-2
- CAL-R
- MPN
- PDGFRA
- PDGFRB
- HLA Typing: For patients fit for AlloSCT
- Chemotherapy consent after informing about disease, prognosis, cost of therapy, side effects, hygiene, food and contraception
- PICC line insertion and Chest X ray after line insertion
- Tumor board meeting and decision
- Attach supportive care drug sheet
- Inform primary care physician
Treatment Plan:
- If there is PDGFR mutation- Imatinib- 400mg-OD
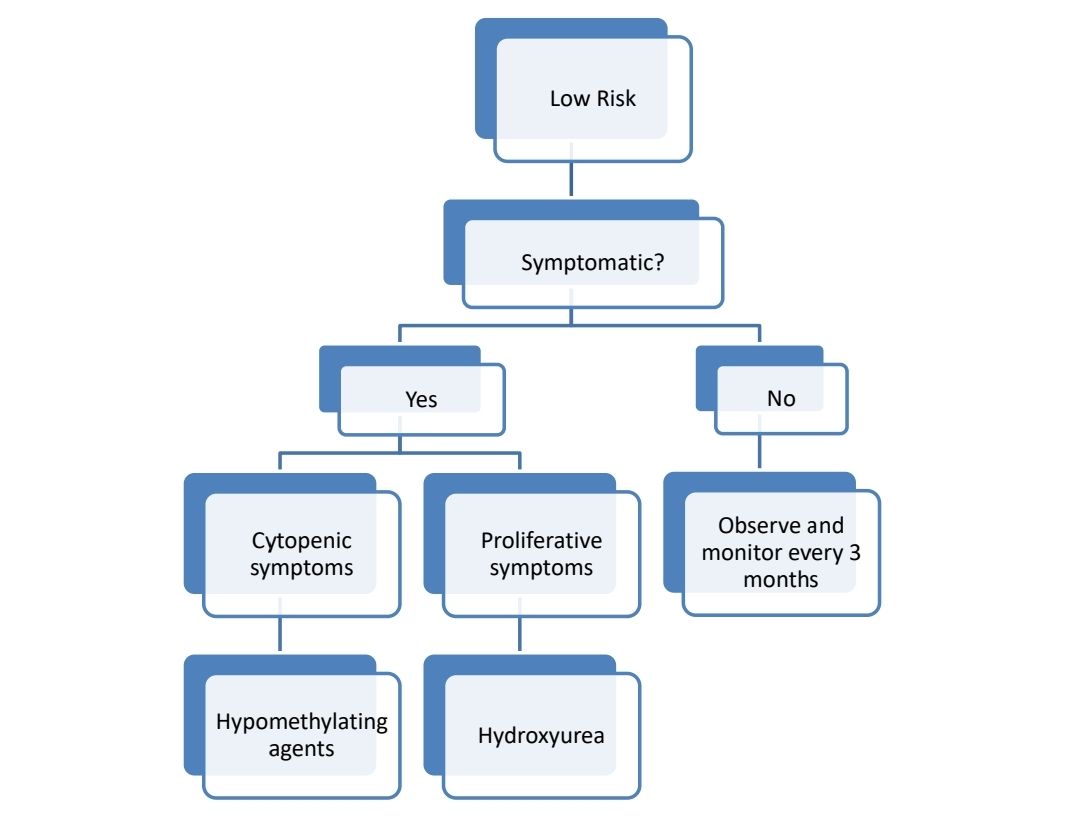
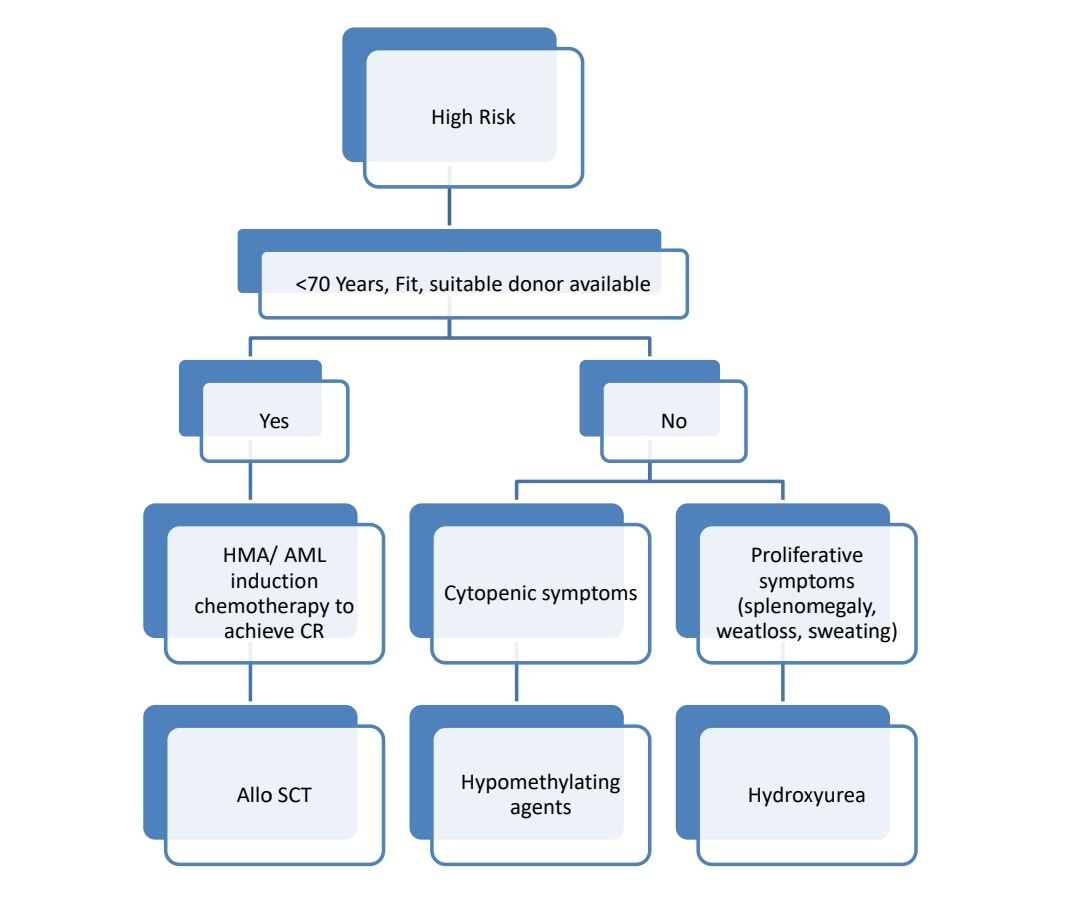
About each modalilty of treatment:
- Allo SCT-
- Only treatment with a chance of cure
- Approximately 1/3rd patients get cured with this approach
- Done in younger patients and patients with good PS and higher risk CMML
- Generally, SCT is done after achieving CR using hypomethylating agents.
- CR with AML like induction therapy may be necessary in CMML-2 with severe cytopenia, rapidly evolving disease, AML-M4 is a close differential diagnosis (due to presence of Auer rods) or presence of NPM1 mutation.
- PBSC is preferred over BM harvest
- Conditioning used include: Busulfan with Cyclophosphamide or TBI based regimens. In case of MUD, ATG is added.
- RIC conditioning may be used in patients with advanced age.
- Hypomethulating agents (Decitabine/ Azacytidine)
- Useful in patients with CMML-2 with WBC count <13,000/cmm, but can be used even in patients with myeloproliferative CMML.
- Dosing same as MDS
- ORR- 40-50%
- Hydroxyurea
- Useful especially in patients with significant leukocytosis or organomegaly with absence of major cytopenias or excess of marrow blasts or renal dysfunction due to high lysozyme levels
- Dose- 500mg- BD, then adjust the dose to target ANC of 500-1000/cmm
- Cytarabine
- Etoposide
- Ruxolitinib- ORR- 38%
- Supportive care including ESA or transfusions
- Steroids:
- Short course may be tried if thrombocytopenia appears to be due to immune destruction
MDS/MPN with neutrophilia
Introduction:
- It is a leukemic disorder that demonstrates both MDS & MPD features
- They have all features of CML but do not have Philadelphia chromosome / BCR-ABL fusion gene
Epidemiology:
- 1-2 cases for every 100 cases of BCR-ABL positive CML
- Common at 70-80 years
Pathogenesis:
- Mutations in ETNK1 and SETBP1 are often seen
Clinical Features:
- Anaemia
- Thrombocytopenia
- Splenomegaly
Investigations:
- Hemogram
- WBC count variable – 35- 96 x 109 /L (Always >13,000/cmm)
- Blasts < 5%
- Immature granulocytes (promyelocyte, myelocyte, metamyelocytes) -10-20%
- Dysgranulopoiesis suggested by acquired Pelger – Huet& Other nuclear abnormalities and abnormal cytoplasmic granularity
- RBC- Macro-ovalocytosis
- Platelet count – Usually thrombocytopenia
- Monocytosis – Up to 10%
- Basophilia
- Bone marrow aspiration and biopsy
- Hypercellularity due to granulocytic proliferation,
- Blasts – Modestly increased in number (But < 20%)
- Dysgranulopoiesis – Present
- Megakaryocytes – Variable count and dysplastic
- Dyserythropoiesis present
- Increased reticulin fibres in some cases.
- Immunophenotyping- No Specific finding
- Cytogenetics and molecular studies
- +13, +8, del (20q), i(17q) & del (12p)
- No BCR/ABL fusion gene
- No PDGFR rearrangement
- Mutations of ASXL1, TET2, and/or DNMT3A are often present
- Other mutations include
- Proliferative signaling pathways: CBL, JAK2, NF1, NRAS
- Splicing machinery: SRSF2, U2AF1, SF3B1, ZRSR2
- Transcription factors: CUX1, GATA2, RUNX1
- Enzymes: ETNK1
- Epigenetic regulators: SETBP1, EZH2
- Chromosomal separation regulators: STAG2
Criteria for Diagnosis:
- Essential
- Peripheral blood leukocytosis ≥13,000/cmm, with neutrophilia and ≥10% circulating immature myeloid cells (promyelocytes, myelocytes and metamyelocytes), as well as neutrophilic dysplasia.
- Hypercellular bone marrow with granulocytic predominance and granulocytic dysplasia, with or without dysplasia in the megakaryocytic and erythroid lineages.
- <20% blasts in blood and bone marrow.
- Not meeting diagnostic criteria for myeloproliferative neoplasms (specifically, exclusion of BCR::ABL1 fusion), myeloid neoplasms with eosinophilia and defining gene rearrangement, chronic myelomonocytic leukemia, or myelodysplastic/myeloproliferative neoplasm with SF3B1 mutation and thrombocytosis.
- Desirable
- Detection of SETBP1 and/or ETNK1 mutations.
- Absence of mutations in JAK2, CALR, MPL, and CSF3R
Prognosis:
- Median survival: 20 months
- 15-40% evolve into AML
- Rest die because of marrow failure
- Poor prognostic markers
- Thrombocytopenia
- Marked anaemia<10gm/dL
- Age >65 years
- Female sex
- WBC- >50,000/cmm
Pretreatment Work-up:
- History
- Examination: Spleen:
- WHO P. S.
- Flow cytometry
- BMA and Bx
- Haemoglobin
- TLC, DLC
- Platelet count
- Peripheral smear
- LFT: Bili- T/D SGPT: SGOT: Albumin: Globulin:
- Creatinine
- Uric acid
- LDH
- HIV
- HBsAg
- HCV
- Cytogenetics
- BCR-ABL1
- JAK 2
- CAL-R
- MPN
- PDGFRA
- PDGFRB
- Chemotherapy consent after informing about disease, prognosis, cost of therapy, side effects, hygiene, food and contraception
- Tumor board meeting and decision
- Attach supportive care drug sheet
- Inform primary care physician
Treatment:
- Allogeneic stem cell transplantation in CR1
- Interferon gamma- Occasional patients achieve complete remission
- Hydroxyurea- useful as palliative treatment.
MDS/MPN with SF3B1 mutation and thrombocytosis
(MDS/MPN with ring sideroblasts and thrombocytosis)
- Median age: 68- 75 years
- Additional mutations often seen include:
- JAK2 p.V617F
- TET2
- DNMT3A
- ASXL1
- SETBP1
- MPL
- SH2B3
- Hemogram
- Macrocytic/ normocytic normochromic anemia
- Thrombocytosis with anisocytosis
- WBC: TLC and DC are often normal
- Blasts: rarely seen
- Bone marrow
- Hypercellular
- Increased erythropoiesis with megaloblastic/ dysplastic features
- >15% ringed sideroblasts are seen on iron staining
- Megakaryocytes are increased and are dysmorphic
- Presents with splenemegaly
- Thrombosis risk is high
- Diagnostic criteria
- Essential
- Anaemia associated with dysplastic erythropoiesis and ≥15% ring sideroblasts, with or without dysplasia in the megakaryocytic and erythroid lineages.
- Persistent thrombocytosis, with platelet count ≥4,50,000/cmm
- SF3B1 mutations or, in the absence of these mutations, concurrent biologically similar mutations involving spliceosome factors and cell signaling
- Not meeting diagnostic criteria for myelodysplastic neoplasms, myeloproliferative neoplasms, chronic myelomonocytic leukaemia, acute myeloid leukaemia with MECOM rearrangement, or myeloid/lymphoid neoplasms with eosinophilia.
- Desirable:
- Concurrent JAK2 p.V617F or other myeloid neoplasm associated mutations
- Essential
- Prognosis:
- Among all MDS/MPN, it has most favorable prognosis
- Median survival: 76- 128 months
Myelodysplastic/myeloproliferative neoplasm, NOS
- It is a myeloid neoplasm with dysplastic and proliferative features that does not meet the criteria for other defined MDS/MPN entities.
- Median age: 70 years
- Clinical features:
- MPN-like: weight loss, night sweats, splenomegaly, and thromboembolic complications
- MDS-like: fatigue, dyspnea, infections, and bleeding, sometimes transfusion dependent anaemia.
- Cytogenetics:
- Abnormal in 50% cases
- Common abnormalities include trisomy 8, monosomy 7/deletion 7q, deletion 20q, and a monosomal or a complex karytoype.
- Molecular studies: TET2, NRAS, RUNX1, CBL, SETBP1 and ASXL1 mutations are common
- Criteria for diagnosis: Essential
- Peripheral blood: Combination of cytopenia(s) and proliferative feature(s)
- Bone marrow cytology: Combination of cell dysplasia and proliferative features
- Molecular analyses of blood or bone marrow: Combination of mutations seen in proliferative and dysplastic myeloid malignancies
- To be excluded
- Therapy-related myeloid neoplasms
- Disease defining gene fusions such as BCR::ABL1 fusion or rearrangement of PDGFRA, PDGFRB, JAK2 or FGFR1
- Biallelic TP53 mutations
- Other MDS/MPN entities, including CMML, MDS/MPN with neutrophilia (MDS/MPNN), or MDS/MPN with SF3B1 mutation and thrombocytosis
- Prognosis: Poor with overall survival of 12- 24 months
Figures:
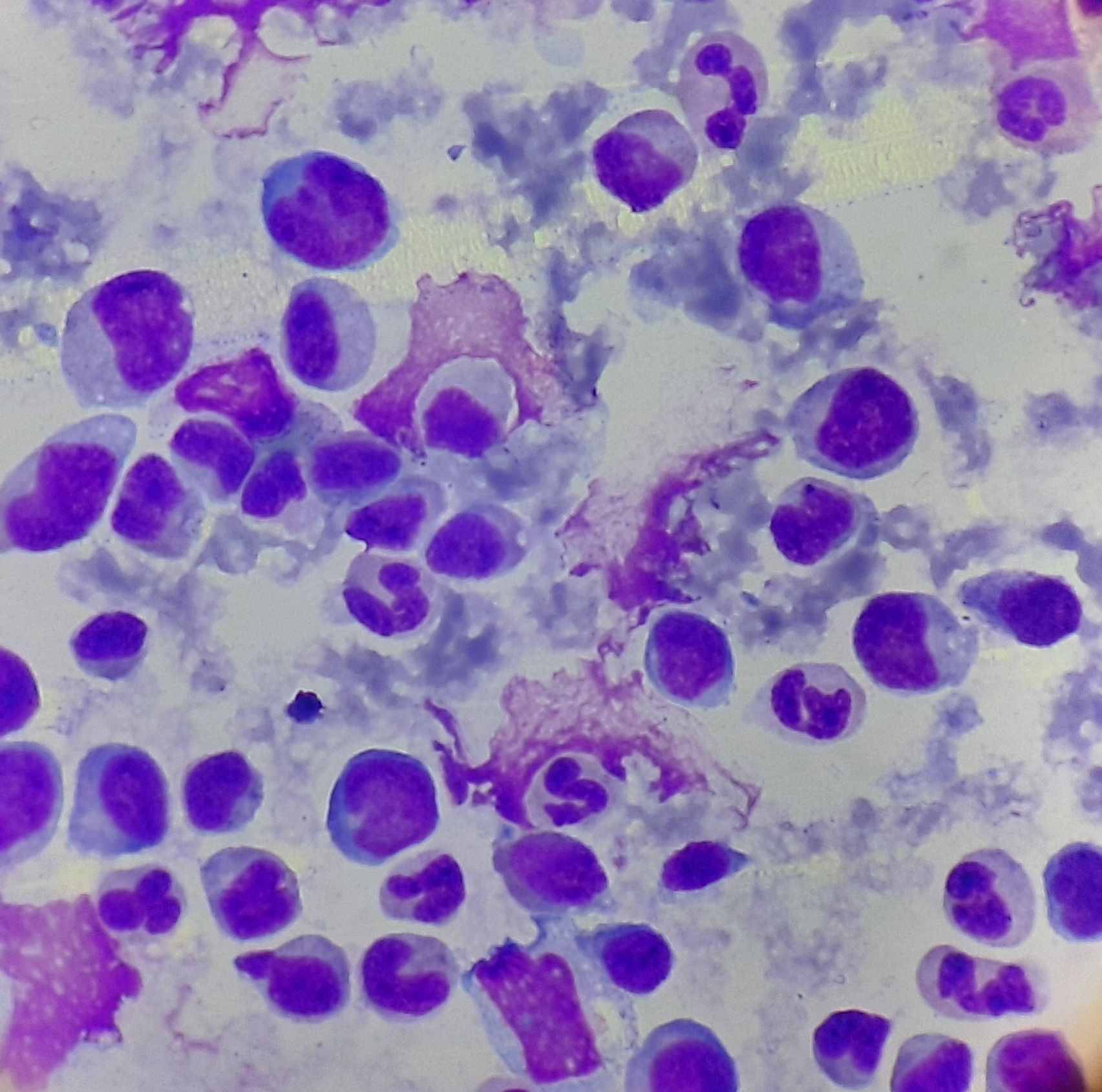
Figure 3.2.1- Chronic myelomonocytic leukemia- Bone marrow aspiration
An Initiative of
Veenadhare Edutech Private Limited
1299, 2nd Floor, Shanta Nivas,
Beside Hotel Swan Inn, Off J.M.Road, Shivajinagar
Pune - 411005
Maharashtra – India

howitreat.in
CIN: U85190PN2022PTC210569
Email: admin@howitreat.in
Disclaimer: Information provided on this website is only for medical education purposes and not intended as medical advice. Although authors have made every effort to provide up-to-date information, the recommendations should not be considered standard of care. Responsibility for patient care resides with the doctors on the basis of their professional license, experience, and knowledge of the individual patient. For full prescribing information, including indications, contraindications, warnings, precautions, and adverse effects, please refer to the approved product label. Neither the authors nor publisher shall be liable or responsible for any loss or adverse effects allegedly arising from any information or suggestion on this website. This website is written for use of healthcare professionals only; hence person other than healthcare workers is advised to refrain from reading the content of this website.