
howitreat.in
A user-friendly, frequently updated reference guide that aligns with international guidelines and protocols.
Hemophagocytic Lymphohistiocytosis
Introduction:
- It is also called as macrophage activation syndrome. Some authorities consider MAS as a separate disease entity which is associated with juvenile rheumatoid arthritis.
- It is an aggressive and potentially fatal syndrome that results from inappropriate prolonged activation of lymphocytes and macrophages.
Epidemiology:
- 1.2/1million children/ year
Etiology:
- Primary/ Familial HLH
- Mutations in any one of following genes
- Perforin- on chromosome 10q21
- h-munc
- Granzyme B
- Syntaxin 11
- GATA2
- LYST (Chediak Higashi syndrome)
- RAB27A (Griscelli syndrome)
- SH2DIA (X linked proliferative disease)
- AP3B1 (Hermansky Pudlak disease)
- Autosomal recessive inheritance
- Bouts of disease are triggered by infections
- Invariably fatal disease with median survival of <2months
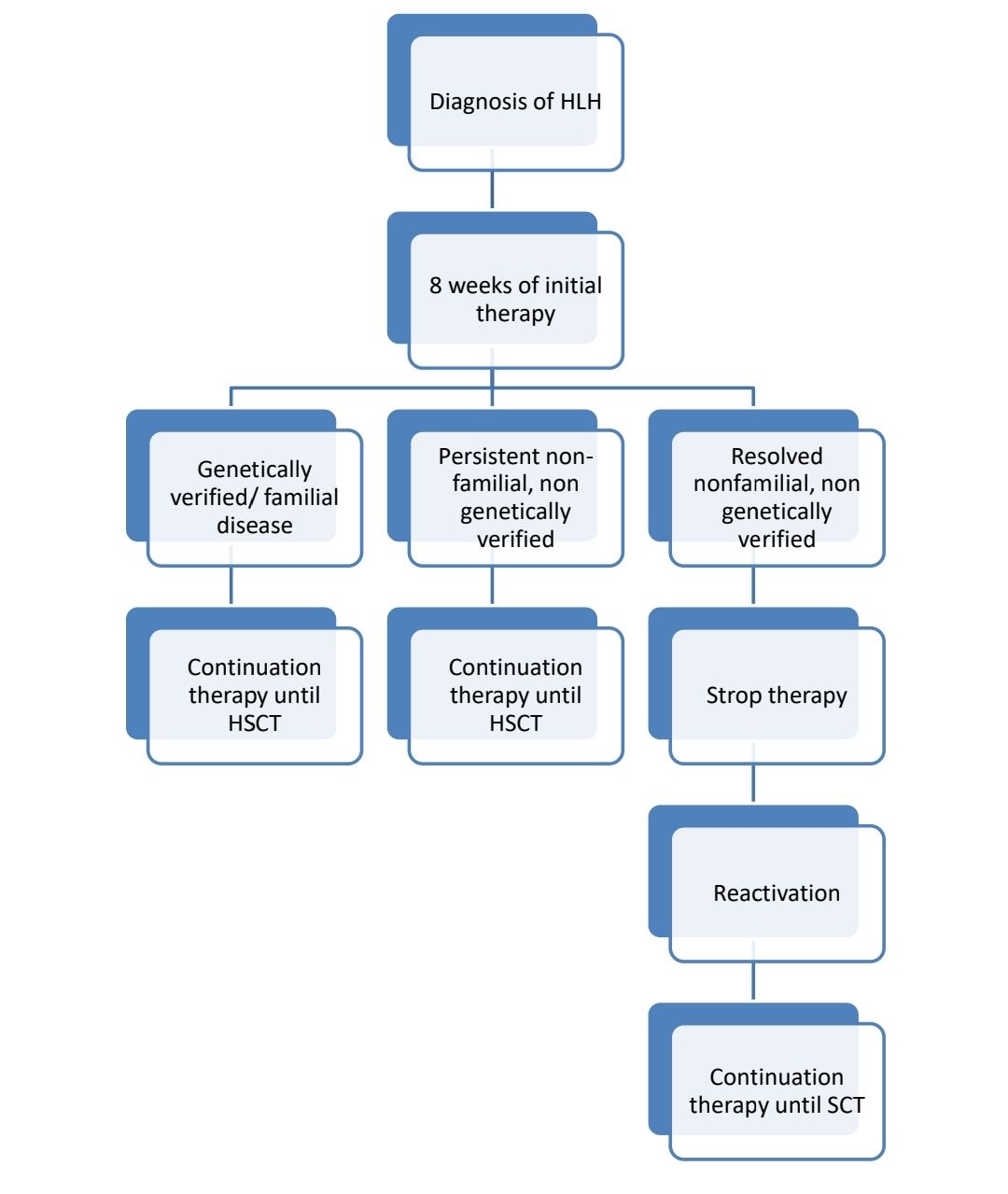
- Need initial/continuation therapy, followed by HSCT
- Mutations in any one of following genes
- Secondary:
- Generally, occurs due to
- Severe infections- Bacteria, mycobacterium, virus (EBV, CMV. HHV-8, HIV, dengue), Parasites
- Immunocompromised host with viral infection
- Autoimmune disorders- Also called as macrophage activation syndrome
- Prolonged IV nutrition
- Leukemia, lymphoma, Solid tumors
- Post splenectomy
- It may subside spontaneously, but is associated with pronounced mortality
- Some have impaired NK cell activity
- Need initial therapy only and treatment of primary disease
- Generally, occurs due to
Pathogenesis:
1.
Defect in the function of NK cells and cytotoxic T cells
↓
Inappropriate activation of T cells and macrophages
↓
Release of proinflammatory cytokines
(Interferon gamma, TNF alpha, IL6, IL10, IL12, soluble IL2 receptor alpha)
↓
Multi-organ dysfunction
↓
Death
2.
Lack of perforin
↓
Lack of killing of macrophages and antigen presenting cells by NK cells
↓
Intense and prolonged systemic inflammation
Clinical Features:
- Fever
- Hepatosplenomegaly
- Cytopenia
- Lymphadenopathy
- Skin rash
- Jaundice
- Edema
- CNS manifestations- Seizures, meningismus, decreased consciousness, cranial nerve palsy, psychomotor retardation, ataxia, irritability, hypotonia
- Family history of HLH
- H/O consanguinity
Investigations:
- Hemogram- Anemia, neutropenia, thrombocytopenia
- S. Triglyceride levels- Elevated
- Coagulation assay- Decreased fibrinogen due to increased plasminogen secretion from macrophages
- LFT- Deranged
- Ferritin- Elevated
- CSF- Pleocytosis
- NK cell cytotoxic activity- Decreased
- BM aspiration
- Hemophagocytosis is seen in 1/3rd of cases
- Accumulation of lymphocytes and mature macrophages is noted
- If not seen, BM can be repeated after few days
- Finding hemophagocytosis is highly suggestive of HLH, but is neither necessary nor sufficient to make the diagnosis
- S. Cytokine levels- Elevated soluble IL-2 receptor, CD163
- Electrolytes- Hyponatremia
- MRI- Discrete lesions, leptomeningial involvement, global edema
- Molecular tests for genetic mutations
- Flow cytometry- To detect presence of perforin
Criteria for Diagnosis:
- If a molecular diagnosis is consistent with HLH, it is enough for diagnosis of HLH
- If molecular diagnosis if negative/ not possible- at least 5 out of 8 criteria have to be present for diagnosis of HLH
- Fever
- Splenomegaly
- Cytopenia affecting >1 cell lineage (Hb <9gm/dL, ANC- <1000/cmm, Platelets <1lac/cmm)
- Fasting Triglyceride >265mg/dL or Fibrinogen <1.5g/L
- Hemophagocytosis in BM or lymph node with no evidence of malignancy
- Low or absent NK cell activity
- Ferritin >500microgm/L
- Soluble CD25 (i.e- soluble IL-2 receptor)- >2400 U/ml
- Diagnostic criteria may not be fulfilled in some patients. But if there is strong clinical suspicion of HLH, therapy should be commenced. Otherwise, overwhelming disease activity may cause irreversible damage.
Criteria for diagnosis of macrophage activation syndrome (Any 2 Lab criteria and any 2 clinical criteria, with hemophagocytosis demonstrated in BM in doubtful cases):
- Laboratory criteria
- Platelet count <2.62lac/cmm
- Increased SGPT- >59 IU/L
- Decreased TLC- <4000/cmm
- Decreased fibrinogen- <250mg/dL
- Clinical criteria
- CNS dysfunction- Irritability, lethargy, disorientation, headache, seizures, coma
- Haemorrhages- Purpura, mucosal bleed
- Hepatomegaly- >3cm below subcostal margin
Prognosis:Predictors of death
- Increased ferritin
- Increased bilirubin
- Decreased albumin
- CSF pleocytosis
- Non-subsiding fever
Differential Diagnosis:
- Langerhan cell histiocytosis
- X linked lymphoproliferative syndrome
- Chediak Higashi syndrome
- Griscelli syndrome
- Lysinuric protein intolerance
- SCID
- Digeorge syndrome with HLH
- Omenn syndrome
- Infections causing pancytopenia
- Rheumatological disorders- Kawasaki disease, SLE, RA
- Malignancies- Lymphoma, leukemia
Pretreatment Work-up:
- History
- Examination
- Haemoglobin
- TLC, DLC
- Platelet count
- Peripheral smear
- Reticulocyte count
- BMA and Bx
- FNA of enlarged LN
- Coagulation Profile: PT: APPT: Fibrinogen:
- LFT: Bili- T/D SGPT: SGOT:Albumin: Globulin:
- Ferritin:
- Fasting TG:
- Soluble IL2 receptors, NK- cell activity- If available
- Molecular test/ Flow for Perforin/ h-munc- if available
- Creatinine
- Electrolytes: Na: K: Ca:Mg: PO4:
- LDH
- S. Immunoglobulins: IgG: IgM: IgA:
- CSF Routine + Cytology
- Inv for infections (PCR)
- CMV: EBV: HSV: HHV6:
- Rubella: Varicella: Parvo: Adeno:
- Leishmania: Brucella: Mycoplasma: TB:
- HIV: HBsAg: HCV:
- Molecular tests
- Perforin: hMunc:
- USG- Abdomen
- Chest X Ray
- MRI- Brain(If CNS Symptoms)
- HLA Typing (For HSCT eligible pt)
- Chemotherapy consent after informing about disease, prognosis, cost of therapy, side effects, hygiene, food and contraception
- Fertility preservation
- PICC line insertion and Chest X ray after line insertion
- Tumor board meeting and decision
- Attach supportive care drug sheet
- Inform primary care physician
Treatment Plan:
- Familial HLH and HLH in less than 18years old- Follow HLH 2004 protocol
- All others- Steroids with treatment of cause initially. If no response, HLH 2004 protocol may be given.
- In addition to HLH treatment, all treatable infections must be treated.
- Underlying malignancies also must be treated.
- Where indicated, HSCT should be performed as early as possible.
- For EBV triggered HLH- Add Rituximab
- If it is macrophage activation syndrome/ HLH due to rheumatological disorders: Initial high dose steroids (Methylprednisolone- 2-3mg/kg/day- in 4 divided doses) followed by Cyclosporine A. If no response treat with HLH protocol containing Etoposide. Etanercept and infliximab have also been found to be useful.
Response Criteria:
- No fever
- Reduction in spleen size
- Platelets >1lac/cmm
- Normal fibrinogen
- Decreasing ferritin levels (by 25%)
About Each Modality of Treatment:
- Initial therapy (for 8 weeks)
- Aims
- Keep the patient alive
- Reduce the number and degree of permanent complications during the initial critical period.
- Achieve resolution of disease
- Inj. Etoposide
- 1st and 2nd week - 150mg/m2- IV- Twice weekly
- 3rd to 8th week- 150mg/m2- IV- Once a week
- Dexamethasone (Oral/IV)
- 1st and 2nd week- 10mg/m2/day
- 3rd and 4th week- 5 mg/m2/day
- 5th and 6th week- 2.5 mg/m2/day
- 7th week 1.25 mg/m2/day
- 8th week- Taper and stop
- Cap. Cyclosporine A
- Start with 3mg/kg- BD- Then adjust the dose to maintain trough levels around 200microgm/L
- Triple IT- 2 doses- 4 weeks apart
- Aims
- Continuation therapy
- Aim: Sustain the resolution of disease
- Inj. Etoposide- 150mg/m2- IV- Every 2nd week
- Tab. Dexamethasone- 10mg/m2 for 3 days- Every 2nd week
- Cap. Cyclosporine A- With target trough levels around 200 microgm/L
- Stem cell transplantation
- If matched sibling donor is not available, haplo identical transplantation has to be considered
- Check whether donor carries HLH mutation
- Conditioning should preferably include- Etoposide, Busulfan, and Cyclophosphamide.
- If unrelated transplant, use ATG.
- GVHD prophylaxis: Cyclosporine and Methotrexate
Supportive Care:
- Broad spectrum antibiotics
- Aggressive transfusion support
- Prophylactic co-trimoxazole
- Oral antimycotic
- Antiviral- Acyclovir
- IVIG- 0.5mg/kg- IV- Once in every 4 weeks (During initial and continuation therapy)
Monitoring After Treatment/ Follow-up:
- Once a month for 3-4 months, then once in 3 months for 2 years
- Monitor- Fever, hepatosplenomegaly, neurological abnormalities
- Hemogram, ferritin, transaminases
Figures:
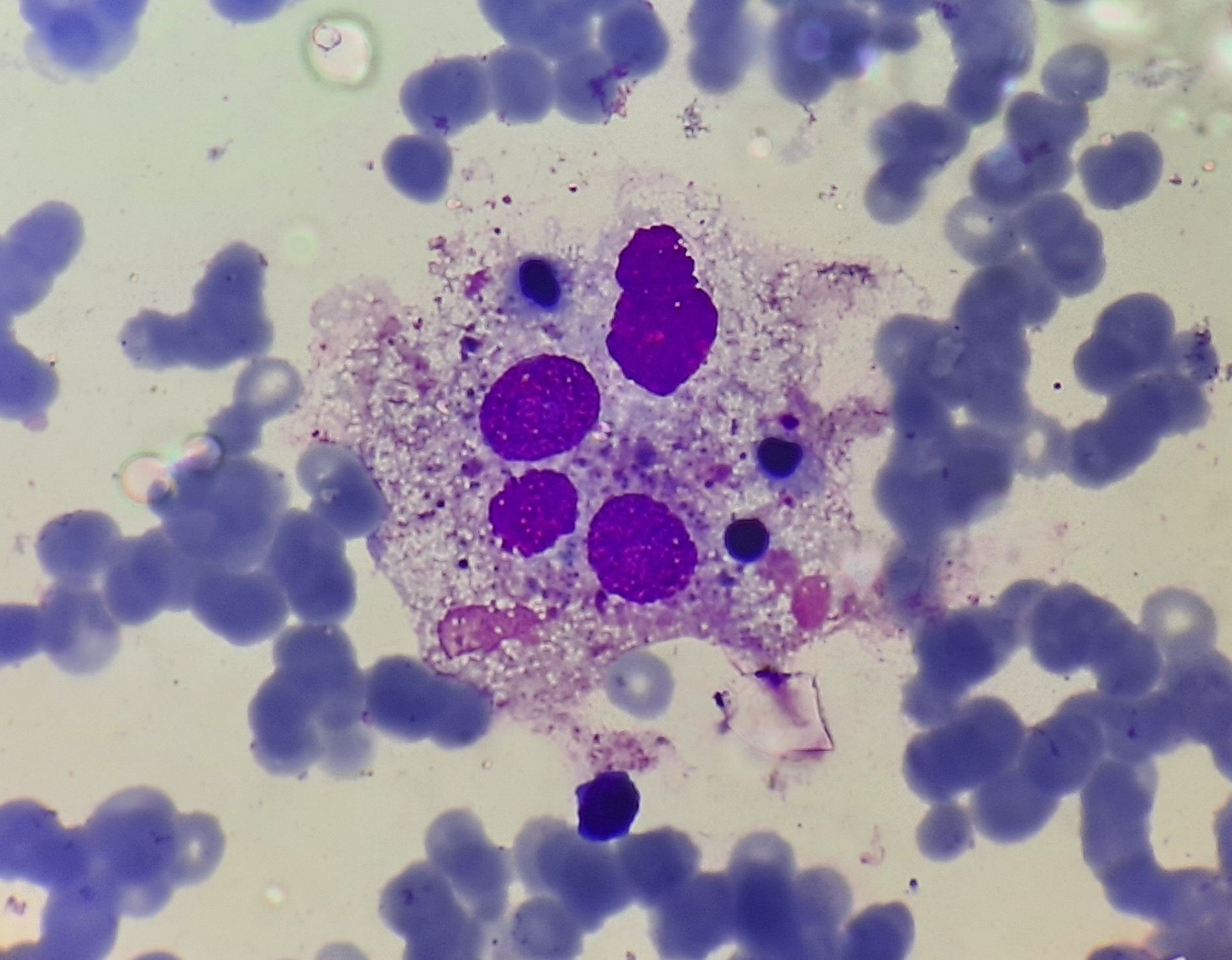
Figure 11.2.1- Hemophagocytosis in bone marrow
Recent advances:
Ruxolitinib-based regimen in children with primary hemophagocytic lymphohistiocytosis
The study aimed to evaluate the effectiveness and safety of a ruxolitinib (RUX)-based regimen as a bridge to hematopoietic stem cell transplantation (HSCT) in children with primary hemophagocytic lymphohistiocytosis (pHLH). Patients received RUX until HSCT or unacceptable toxic side-effects, with methylprednisolone and etoposide added sequentially if the disease was suboptimally controlled. The primary endpoint was 1-year overall survival. Results showed that 90.5% of patients achieved a complete response within the first 8 weeks, and 81.0% were alive at the last follow-up, with a 1-year overall survival of 90.5%. Most patients tolerated the RUX-based regimen well, with hematologic adverse events being the most frequently observed.
https://doi.org/10.3324/haematol.2023.283478
Etoposide improves survival in primary hemophagocytic lymphohistiocytosis
In a study of 88 patients with primary hemophagocyticlymphohistiocytosis (pHLH) from 2016 to 2021, first-line etoposide-based therapy was administered to 86% of symptomatic patients, leading to improved survival rates compared to previous studies. Hematopoietic stem cell transplantation (HSCT) was performed in 75 patients, with a 3-year probability of survival of 82% for the entire cohort and 77% for those receiving first-line etoposide. Survival rates pre- and post-HSCT improved compared to previous studies, attributed to reduced-toxicity conditioning and shorter time from diagnosis to HSCT. Notably, early HSCT for asymptomatic patients resulted in 100% survival, highlighting the potential benefit of newborn screening for pHLH.
https://doi.org/10.1182/blood.2023022281
The effectiveness of the doxorubicin-etoposide-methylprednisolone regimen for adult HLH secondary to rheumatic disease
This retrospective study evaluated the efficacy of the doxorubicin-etoposide-methylprednisolone (DEP) regimen in 58 adult patients with hemophagocytic lymphohistiocytosis (HLH) secondary to rheumatic disease. The overall response rate was 82.8%, with 13.8% achieving complete response and 69% partial response. Serum markers such as ferritin, sCD25, ALT, AST, and DBIL significantly decreased over time. The mortality rate was 20.7%, and poor prognosis factors included advanced age, low hemoglobin and platelet counts, and CNS involvement. The DEP regimen showed high efficacy with acceptable side effects in this patient population.
https://doi.org/10.1007/s00277-024-05796-8
Emapalumab therapy for hemophagocytic lymphohistiocytosis before reduced-intensity transplantation improves chimerism
This retrospective study highlights the benefits of emapalumab, an anti–IFN-γ antibody, in improving post-HSCT outcomes for pediatric hemophagocytic lymphohistiocytosis (HLH) patients undergoing reduced-intensity conditioning (RIC) HSCT. Among 50 patients, emapalumab use within 21 days before conditioning was associated with significantly lower rates of mixed chimerism (48% vs. 77%) and severe mixed chimerism (5% vs. 38%). Furthermore, intervention-free survival (IFS) was significantly improved with emapalumab (73% vs. 43%), particularly in infants aged <12 months, a high-risk group (75% vs. 20%). While overall survival was higher in the emapalumab group (82% vs. 71%), the difference was not statistically significant. These findings suggest that emapalumab effectively reduces graft failure risk and enhances IFS, making it a valuable adjunct in managing HLH before RIC-HSCT.
https://doi.org/10.1182/blood.2024025977
An Initiative of
Veenadhare Edutech Private Limited
1299, 2nd Floor, Shanta Nivas,
Beside Hotel Swan Inn, Off J.M.Road, Shivajinagar
Pune - 411005
Maharashtra – India

howitreat.in
CIN: U85190PN2022PTC210569
Email: admin@howitreat.in
Disclaimer: Information provided on this website is only for medical education purposes and not intended as medical advice. Although authors have made every effort to provide up-to-date information, the recommendations should not be considered standard of care. Responsibility for patient care resides with the doctors on the basis of their professional license, experience, and knowledge of the individual patient. For full prescribing information, including indications, contraindications, warnings, precautions, and adverse effects, please refer to the approved product label. Neither the authors nor publisher shall be liable or responsible for any loss or adverse effects allegedly arising from any information or suggestion on this website. This website is written for use of healthcare professionals only; hence person other than healthcare workers is advised to refrain from reading the content of this website.