
howitreat.in
A user-friendly, frequently updated reference guide that aligns with international guidelines and protocols.
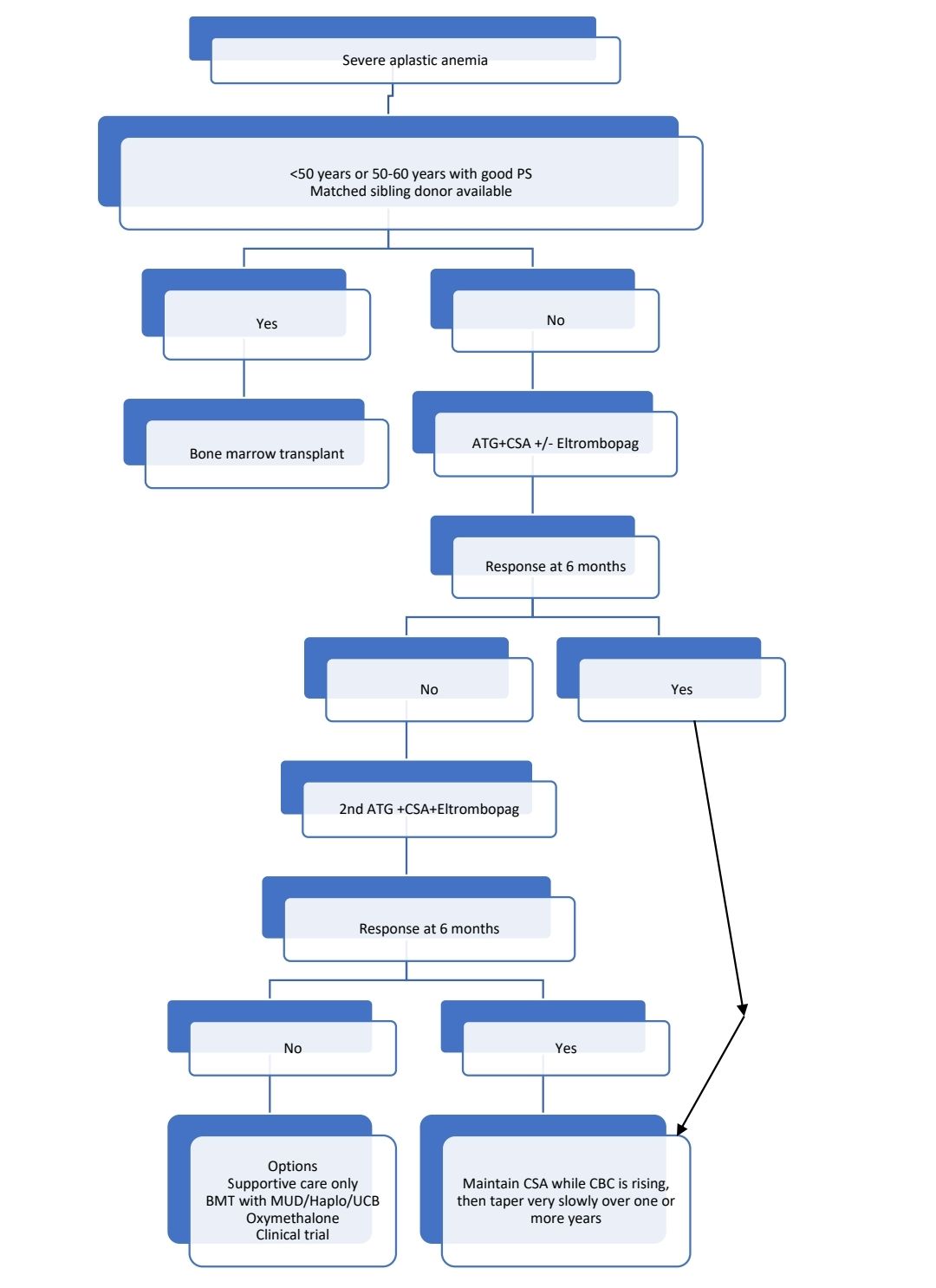
Idiopathic Acquired Aplastic Anemia
Introduction:
- It is a disorder characterized by pancytopenia resulting from aplasia of bone marrow in the absence of abnormal infiltrate and with no increase in reticulin.
Epidemiology:
- Incidence- 0.6-6/million population
- No race/age/ sex predilection
- High incidence in Asia
- 50% of cases occur before 3rd decade
- Biphasic age peak- 1-25years and more than 60 years
- In India 6000-8000 patients/ year
Etiology:
- “Seed, soil, worm, and fertilizer hypothesis”
- Seed- Functioning hematopoietic stem cells
- Soil- Bone marrow micro environment
- Worm- cellular/humoral immunosuppression
- Fertilizer- Deficiency of growth factor
- ? Primary stem cell defect: Colony cultures have shown that stem cells are unresponsive to even high doses of growth factors
- ? Problem with microenvironment which is less supportive to stem cells- Hence BM harvest is better than peripheral stem cell harvest when transplant is being done
- ? Increased telomere shortening – Patients with aplastic anemia show increased TRF loss in leucocytes compared with age matched control subjects.
- ? Immune mediated apoptosis of stem cells
- Role of immune dysfunction was suggested when autologous transplant recovery was documented in patients with aplastic anemia in whom engrafting failed after bone marrow transplant. Improvement was because of immunosuppression given for conditioning
- Increased association of aplastic anemia with HLA DR-2, especially DR-15 split has been noted
- Regulatory T cells (T regs) are decreased in aplastic anemia. Their number increases with hematological response.
- PNH is sometimes associated with aplastic anemia
- Lack of PIGA leads to increased sensitivity of hematopoietic cells to complement lysis.
- 20% of aplastic anemia patients have evidence of PNH
- They show good response to immunosuppressive therapy
Pathogenesis:
? Infection/ ?Toxin/ ?Drug/ ?Autoimmune reaction/ ?Unknown antigen
↓
Constitutive expression of T bet (transcriptional regulator that is critical to Th1 polarization and INF-gamma production)
↓
Predominant TH 1 cell immune response with release of INF-gamma, IL-2, TNF- α
↓
Increased expression of perforin on activated cytotoxic T lymphocytes and NK cells
↓
Induction of FAS expression on CD34+ Cells
↓
Apoptotic cell death
Clinical Features:
- Petechiae and ecchymoses due to thrombocytopenia (most common)
- Frequent infections due to granulocytopenia- Pneumonia and septicemia are frequent cause of death
- Anemia- Fatigue, lassitude, shortness of breath, ringing in the ears etc
- Stigmata of inherited marrow failure syndromes: skin pigmentation, short stature, microcephaly, hypogonadism, mental retardation, skeletal anomalies, oral cavity
- Lymphadenopathy and organomegaly are absent. Their presence suggest alternative diagnosis
- Get thorough history for identifying secondary causes of aplastic anemia
Investigations:
- Hemogram
- Pancytopenia
- Normocytic normochromic RBCs. Mild macrocytosis is usually observed
- Leucopenia especially neutropenia
- No abnormal cells are seen
- Platelets – Reduced counts, usually small in size
- Bone marrow aspiration
- Can be dry tap
- Fragments are usually easily obtained and they are hypocellular.
- Erythropoiesis is suppressed. Marked dyserythropoiesis is common. So this alone does make the diagnosis of MDS.
- Granulocytes and megakaryocytes are decreased or absent. Presence of dysplasia in them indicates diagnosis of hypocellular MDS.
- Residual lymphocytes, plasma cells, mast cells and macrophages are seen.
- Marrow lymphocytes >75% correlates with poor prognosis
- Trephine biopsy
- Should be minimum 2cm long
- Avoid tangential biopsies, as subcortical marrow is normally hypocellular
- Hypocellular marrow
- <30% in age <60 years
- <20% in age >60years
- Empty marrow spaces populated by fat cells, fibrous stroma and scattered lymphocytes and plasma cells.
- Very few precursor cells are seen (<25%)
- Megakaryocytes are usually absent
- Hot spots: Focal hyperplasia of erythroid or granulocytic cells in different stages of maturation may be seen
- Sometimes lymphoid aggregates are seen, especially if aplastic anemia is associated with systemic autoimmune disorder such as SLE or rheumatoid arthritis
- In a bone marrow of aplastic anemia, always look for hypocellular MDS, hypocellular AML, hairy cell leukemia, lymphoma, myelofibrosis and mycobacterial infection
- ESR – Elevated
- Reticulocyte count – Decreased
- Absolute reticulocyte count - <40x109/L
- Corrected count - <1%
- Flow cytometry for CD34 cells- Decreased
- Radioactive iron surface counting
- Most of iron is taken up by liver rather than bone marrow
- Serum erythropoietin level –Elevated
- Hemoglobin electrophoresis: Hb F content is increased up to 1.5g/dL
- Serum iron content – Increased
- Flow cytometry for CD 55 & CD 59- To rule out PNH. To be done at diagnosis, 6 monthly after treatment for 2 years, and then annually.
- Cytogenetics
- Difficult to get metaphases due to hypocellular marrow
- By FISH karyotyping/ by Single nucleotide polymorphism array (SNP-A) 19% patients show chromosomal abnormalities
- Chromosome 7 abnormalities especially Monosomy 7 (Most common)
- Others: Trisomy 8, 5q deletion, deletion 20q, deletion 13q, trisomy 1
- Trisomy 8 and del 13q herald good prognosis
- Presence of cytogenetic abnormality does not rule out the diagnosis of aplastic anemia
- Chromosome breakage study using diepoxybutane, or mitomycin C (stress cytogenetics)
- Positive in Fanconi’s anemia
- It should be done in all patients of less than 35 years presenting with aplastic anemia
- Serological tests for HIV, EBV, CMV, hepatitis (A/B/C), parvovirus B19
- Measurement of peripheral blood leucocyte telomere length- By PCR or flow FISH methods
- Next Generation Sequencing:
- 54%- Non pathogenic gene mutation, 31%- Classic bone marrow failure genes like FANC, 14%- Primary immunodeficiency associated genes
- Somatic mutations commonly seen- TP53, ASXL-1, DNMT3, RUNX1 (these cases are associated with poor prognosis)
- Mutations typical of myeloid malignancies help to differentiate aplastic anemia from hypocellular MDS.
- Finding of DNMT3A or ASXL 1 mutation does not alter the diagnosis of aplastic anemia.
- Mutations in PIGA, BCOR and BCORL1 genes (these cases are associated with good prognosis)
- Tests for specific mutation causing telomere shortening. Done only if there is short telomere.
- Vitamin B12 levels and folate levels: As some megaloblastic anemia can present with hypocellular bone marrow
- LFT: To detect antecedent hepatitis
- ANA and anti-DsDNA
- DCT and ICT
- Chest X ray: To exclude infections and for comparison with subsequent films
- X ray radii- In young patients, to exclude Fanconi anemia
- Abdominal USG- Enlarged lymph nodes and splenomegaly indicate possibility of malignant hematological disorder
- MRI
- To distinguish marrow fat and hematopoietic cells
- T1 weighted spin echo images
- Fatty marrow appears bright
- Cellular marrow exhibits low density signals
- HLA typing- To identify potential donor, especially in case of young patient
- Flow cytometry for CD57 cells, if many large granular lymphocytes are seen
Criteria for Diagnosis:
- Bone marrow - <25% cellularity without presence of abnormal cells or fibrosis and any two of following findings
- Granulocytes - <1.5x109/L or absolute neutrophil count <0.5x109/L
- Platelet - <50x109/L
- Corrected retic count - <1%
- Hb<10g/dL
Grading of severity: (Camitta criteria)
- Severe aplastic anemia: Bone marrow cellularity<25% with at least two of following 3 criteria
- Absolute neutrophil count <500cells/cmm
- Platelet count <20,000/cmm
- Reticulocyte count <20,000/cmm or <1%
- Very severe aplastic anemia: Same as severe aplastic anemia but absolute neutrophil count is <200/cmm
- Non severe aplastic anemia: Aplastic anemia not fulfilling criteria of severe/ very severe aplastic anemia
Prognosis:
- Untreated patients: 70% die within 5 years of diagnosis
- 20% patients spontaneously recover with supportive therapy
- With immunosuppressive therapy- 5 year survival rate is 75%
- With bone marrow transplant from matched donor- 5 year survival is>90%
- Those due to exposure to toxin, have better prognosis than idiopathic ones.
- Major causes of death:
- Infections
- Bleeding
- In BMT patients: GVHD, toxicity from conditioning regimen
- In immunosuppressant responders: PNH, MDS, Leukemia
Pretreatment Work-up:
- History
- Drugs/Chemicals
- Family history
- T-GVHD
- Pregnancy
- Transfusions
- Examination
- Stigmata of IBMFS
- Hemoglobin
- TLC, DLC
- ANC:
- Platelet count
- Peripheral smear
- Reticulocyte count
- HbF %
- BMA and BxLook for dysplastic features/ Blasts/ Hairy cells
- Reticulin stain on BM Biopsy
- Cytogenetics
- PB- Chromosomal Breakage analysis (For <50 years)
- PNH by Flow
- Nutritional evaluation
- S. B12 level:
- S. Folate level:
- LFT: Bili- T/D SGPT: SGOT:Albumin: Globulin:
- Creatinine
- Electrolytes: Na: K:
- Uric acid:
- LDH
- HIV:
- HBsAg:
- HCV:
- Other viral studies
- HAV: EBV: CMV: Parvo:
- ANA and ANtiDs DNA
- UPT/ Beta HCG
- USG Abdomen
- Chest X Ray
- ECHO (In young pt)
- Telomere length(If available)
- PICC/Hickman insertion
- Sperm preservation (If undergoing HSCT)
Treatment Plan:
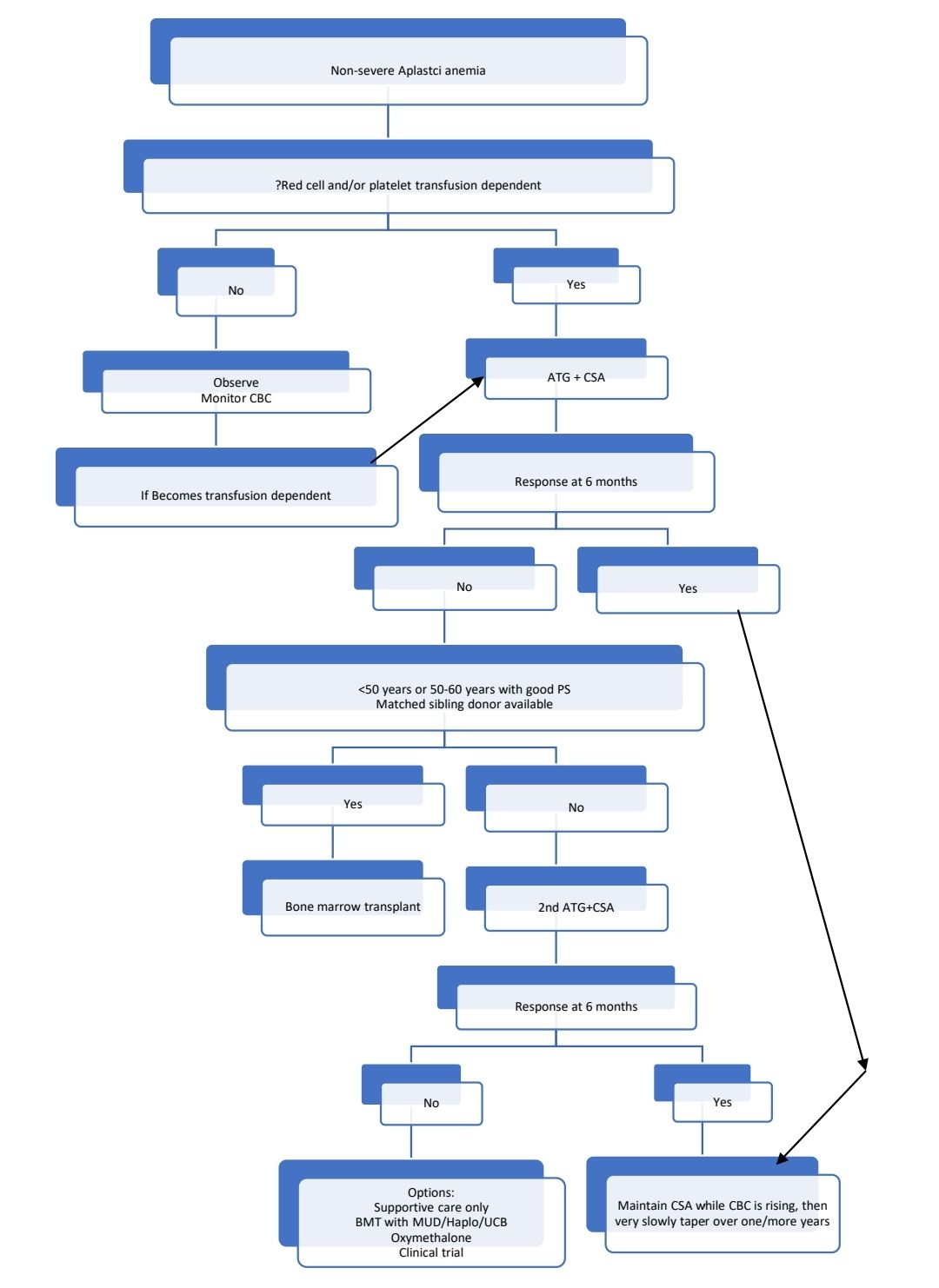
Poor patients who cannot afford ATG or BMT
- Cyclosporine A + Danazol +/- Eltrombopag
Response Criteria:
- Complete response:
- Hemoglobin concentration normal for age and gender
- Neutrophil count- >1500/cmm
- Platelet count- >1,50,000/cmm
- Partial response:
- Transfusion independence
- No longer meet the criteria of severe aplastic anemia
Treatment:
- It should be considered as medical emergency, as lives are lost, if grave consequences of pancytopenia go unrecognized
- Bleeding and infections must be controlled before starting transplant or immunosuppression (Except in cases with fungal infections, where early neutrophil recovery by BMT may help to salvage the patient)
- In patients in whom BMT may be attempted, use transfusions very judiciously, as minimally transfused patients achieve better results.
- Routine bacterial/ viral/ PCP prophylaxis is not necessary.
- Transfusion support:
- Blood products must be leucoreduced (to prevent alloimmunization and CMV transmission) and irradiated (to prevent alloimmunization and transfusion associated GVHD)
- Threshold hemoglobin levels must be individualized
- Rh and Kell matched blood must be used
- Routine platelet transfusions are not necessary for stable patients, who are not on any active treatment.
- Treatment of cause if found:
- Post-transfusion GVHD- Uniformly fatal
- Pregnancy:
- Often association is coincidental.
- 50% spontaneously resolve after delivery/ MTP.
- Transfusions must be given to maintain hemoglobin >8gm/dL and platelet count >20,000/cmm
- Consider MTP if feasible
- G-CSF may be used
- In select cases prednisolone +/- Cyclosporine may be used
- If aplastic anemia persists after delivery- ATG/ BMT should be considered.
- Post hepatitis aplastic anemia: Occurs usually because of non-ABC virus. Very poor prognosis. Mortality- 90% at 1 yr. History of hepatitis is an indication for early BMT
- Post mononucleosis aplastic anemia- Some recover spontaneously
- Collagen vascular disease- Aplastic anemia seen with eosinophilic fasciitis has very poor prognosis
- Any offending drugs- Stop them immediately
About Each Modality of Treatment:
- BMT from HLA identical sibling/ Syngeneic donor
- Best to do it within 180 days of diagnosis
- For Fanconi anemia and other inherited BM failure syndromes, different conditioning regimens are used.
- Conditioning:
- High dose cyclophosphamide- 50mg/kg/day for 4 days (Days -5 to -2)
- 12 hours after the first, second, and third dose of Cyclophosphamide, horse ATG (Atgam) at 30 mg/kg i.v. per dose infused over a period of 10 to 12 hours.i.e. 3 days (Days -5 to -3). Transplant without ATG is also possible.
- Each vial of ATGAM contains 250mg of ATG
- Prior to infusion test dose has to be given
- Methyl prednisolone- 2mg/kg for 3 days (Days -5 to -3)
- Although this regimen is not myeloablative, the immunosuppression is sufficient to allow engraftment in most of the cases.
- Unmanipulated bone marrow is administered 36 hours after the last dose of cyclophosphamide (Day 0).
- Avoid use of peripheral blood stem cells as they produce unacceptably high risk of GVHD. In contrast to malignancies, in case of aplastic anemia there is absolutely no need of GVHD.
- Dose of stem cells- 3x108 mononuclear cells/kg or 3x106 CD34+ cells/kg
- GVHD prophylaxis
- Ciclosporine- 2.5mg/kg- BD. Start from day -1 and continue for 12 months. Tapering to be started from at 9 months to help prevent late graft failure. Trough levels- Adults- 250-350 microg/L and children- 15-200 microgm/L
- Short course of methotrexate- 15mg/m2 on day +1 and 10mg/m2 on days +3, +6 and +11
- Avoid radiation based conditioning due high transplantation related long-term morbidity and mortality
- Use of fludarabine is associated with high risk of CMV reactivation
- Survival is better if donor is of same sex.
- Male patient with female donor- High risk of GVHD
- Female patient with male donor- High risk of rejection
- Overall survival is 75-90%
- Post-transplant monitoring should be done as any other HSCT. Refer to HSCT section. Avoid vaccinations as they may trigger relapse of aplastic anemia.
- Progressive mixed chimerism (>10% recipient cells or >15% increase over 3 months) is associated with high risk of late graft failure. In such condition CSA should not be tapered.
- BMT from haploidentical donor
- Conditioning with EBMT SAAWP protocol
- High risk of graft rejection and severe GVHD
- It should be considered as experimental therapy
- HSCT from unrelated cord blood
- Conditioning with EBMT adopted French protocol of Fludarabine, cyclophosphamide with ATG with 2 Gy TBI with Rituximab on Day +5
- Total nucleated cell dose should be >4x107 cells//kg. Hence often 2 units are required for an adult patient.
- Cord blood lymphocytes are naive and allotransplantation with HLA disparity is feasible without significantly higher risk of GVHD.
- 3 Year overall survival- 38%
- Antithymocyte globulin (Horse ATG) with Ciclosporine A
- Horse ATG: 40mg/kg/day in 500ml NS over 12-18 hours for 4 days. (Each vial contains 250mg)
- On first day- Add only 1 vial to 500ml NS and give slowly over 30min. If no reaction, add remaining vials.
- Each daily dose should be preceded by IV Methylprednisolone (1-2mg/Kg), chlorpheniramine maleate and paracetamol- Minimum 30min before starting Horse ATG.
- It is a crude drug with antibodies against multiple antigens
- Highly immunosuppressive therapy, hence isolation of patient with reverse barrier nursing is necessary. All principles of febrile neutropenia need to be followed.
- High risk of serum sickness- Hence prophylactic steroids have to be administered (Prednisolone 1mg/kg for 2 weeks, then taper slowly)
- Should be given through central line
- If there is fever/ rigor/ rash/ hypotension- Slowdown the rate of infusion, give corticosteroids, antihistaminics and pethidine
- Frequent platelet transfusions are necessary to maintain platelet count >30,000/cmm
- Irradiated blood products must be given to patients receiving ATG and it should be continued indefinitely or at least till absolute lymphocyte count is >1000/cmm
- Generally, 3-4 months are required for improvement of marrow function. Till that time supportive transfusions have to be given.
- Rabbit ATG has very poor response rate and overall survival, hence is not used.
- Ciclosporine
- 2.5mg/kg BD
- Given from day 5 of ATG
- To be taken on empty stomach with fruit juice.
- Plenty of fluids should be advised to maintain adequate urine output.
- After achieving maximal hematological response/ minimum of 1 year, taper the dose very slowly (25mg every 3 monthly)
- Monitor BP, RFT, LFT, and cyclosporine levels
- Target trough levels- Adults- 150-200microgm/L, Children- 100-150 microgm/L
- 65% patients respond to this therapy. 1/3rd of them have relapse.
- Factors indicating goods response:
- Young age
- Less severe disease
- Trisomy8/ del (13q)
- Presence of PNH clone
- Long telomeres
- Overall5-year survival- 75%
- Other immunosuppressives that have been used
- Alemtuzumab- Response rate is 35%-55%. Total dose 100mg- Given as subcutaneous dose 10mg on day 1 and 30mg on days 2,3 and 4.
- Mycophenolate mofetil
- High dose Cyclophosphamide: 40-50mg/kg/day for 4 days. Good response rates are claimed. Prolonged neutropenia limits its widespread use.
- Eltrombopag
- Useful in high doses- start at dose of 50mg/day and increase dose by 50mg every 2 weeks until platelet count is >50,000/cmm. Maximun dose of 150mg/day
- Overall response rate-30%
- Often added to standard immunosuppressive therapy (In such case overall response rate increases to >80%)
- In patients with trilineage response/ transfusion independence for more than 8 weeks dose can be reduced by 50%. Stop once at lowest dose they fulfill above criteria for 8 weeks. If ANC<500/cmm, platelet count <30,000/cmm, Hemoglobin <9gm/dL, restart at the last lowest dose.
- If no response at 16 weeks, it should be stopped.
- It stimulates cMPL receptors in stem cells, hence increases number of bone marrow CD34+ cells.
- Possibility of clonal evolution exists. But it can be used in patients with cytogenetic abnormalities and MDS related mutations. But regular monitoring for new cytogenetic abnormality should be done.
- Androgens:
- Options: Danazol and Oxymetholone- 2.5mg/kg/day
- Increase telomerase activity via aromatization of estradiol to steroids
- If no response after 6 months, it should be stopped
- Has 10-20% chance of response
Note:
- Never use steroids. They are ineffective in treatment of aplastic anemia and they encourage bacterial and fungal infections. They can precipitate serious GI hemorrhage in presence of severe thrombocytopenia.
- G-CSF is not useful in aplastic anemia, as its level is markedly elevated in most of the patients.
- Erythropoietin is also not useful, as its level is also markedly elevated. With cyclosporine, it can cause marked rise in BP.
Figures:
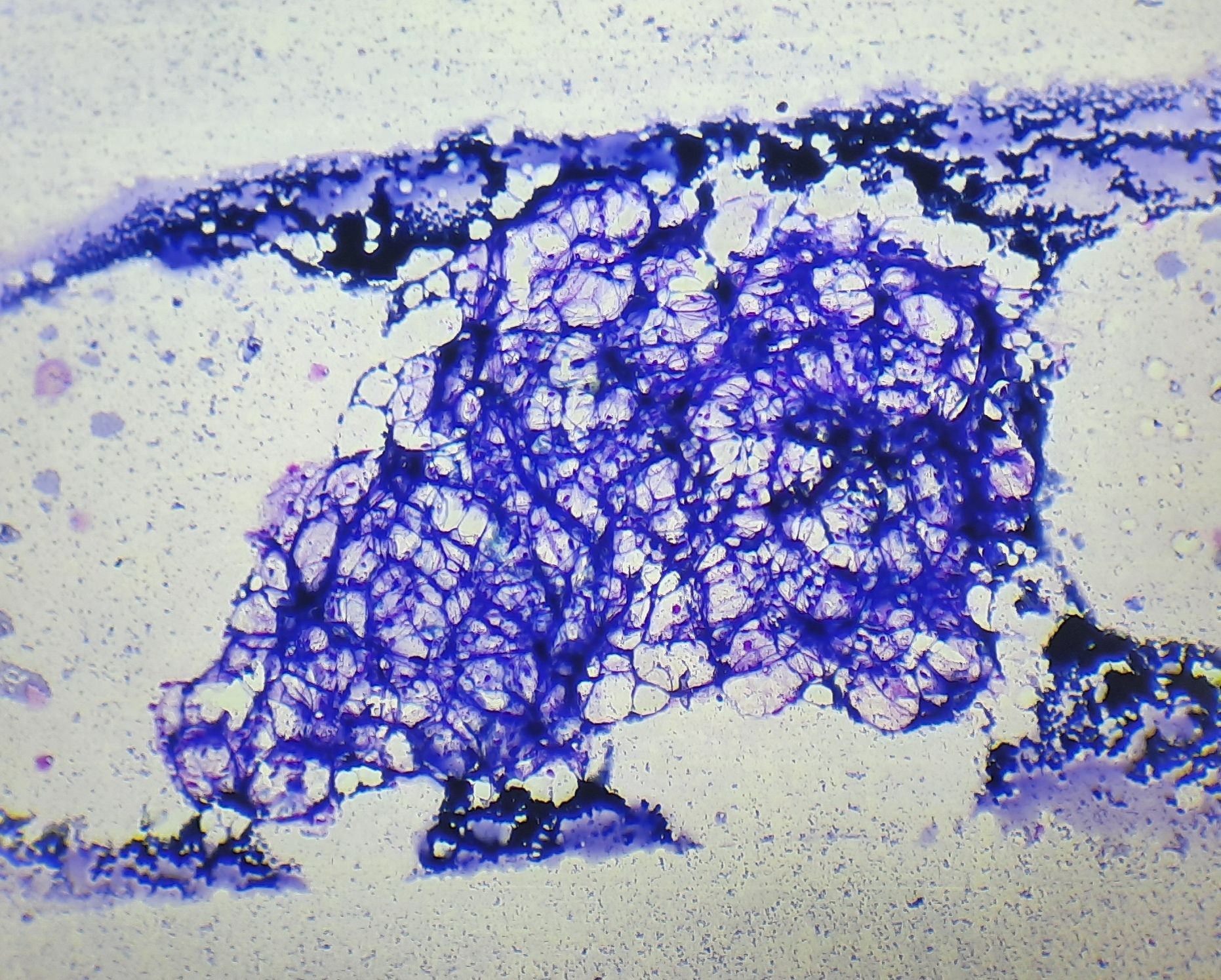
Figure 8.4.1- Aplastic anemia- Bone marrow aspiration- Hypocellular BM fragment
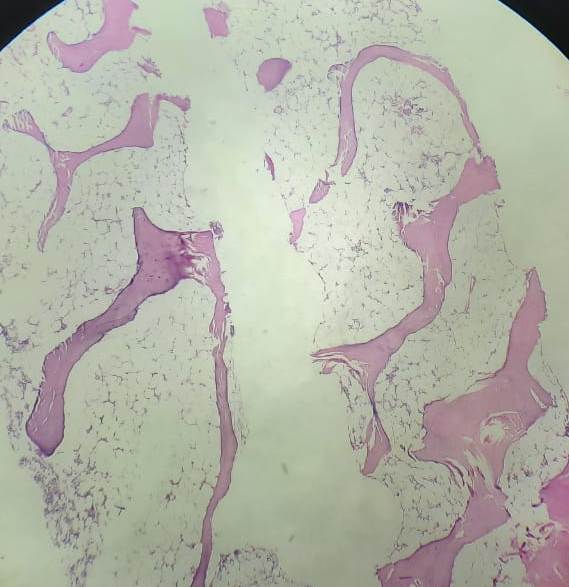
Figure 8.4.2- Aplastic anemia- Bone marrow biopsy- Hypocellular bone marrow
Recent advances:
Inherited human Apollo deficiency causes severe bone marrow failure and developmental defects
Apollo is a genome caretaker critical for the proper development of the immune-hematological system in humans. A recent study by LaëtitiaKermasson et al, showed that patients with severe dyskeratosis congenita had mutation of gene encoding the 5′-to-3′ DNA exonuclease Apollo/SNM1B. Apollo-deficient cells from patients exhibited spontaneous chromosome instability. Patients with this mutation had early-onset hypocellular bone marrow failure, B and NK lymphopenia, developmental anomalies, microcephaly, and/or intrauterine growth retardation.
doi.org/10.1182/blood.2021010791
Cancers after HLA-matched related bone marrow transplantation for aplastic anemia
A recent study analyzed subsequent cancers in patients with aplastic anemia who underwent Allo-HSCT. Study included total of 329 patients. 53 cancers occurred in 46 patients, 42 had solid tumors and 4 blood cancers. The 26-year cumulative incidence (CI) of cancer was 11% and mortality was 5%.
https://doi.org/10.1038/s41409-021-01498-1
Eltrombopag enhances response rate when combined to standard IST
In a recent study from NIH, Patel et al evaluated role of Eltrombopag in treatment of severe aplastic anemia. They had administedEltrombopag alongside horse ATG and Cyclosporine. At 6 months, 81% of patients treated with this combination achieved haematological response compared with 67% of historical controls treated with IST alone. Follow up these patients showed that, haematological responses were not maintained after eltrombopag was discontinued. 39% of patients relapsed. The event-free overall response rate in eltrombopag cohort was similar to historical controls (56% Vs 57%).
https://doi.org/10.1182/blood.2021012130
Germ line mutations with predisposition to myeloid neoplasms in adults with marrow hypocellularity
This study investigated germ line genetic predisposition to myeloid neoplasms in adult patients with hypoplastic bone marrow. Using targeted sequencing, 6.7% of the 402 patients were found to carry germ line variants linked to predisposition syndromes. Conditions included DDX41-associated predisposition, Fanconi anemia, GATA2-deficiency syndrome, and others. Of those with causative germ line mutations, 67% were diagnosed with myeloid neoplasms. Patients with predisposition variants were typically younger, had a higher risk of severe cytopenias, and advanced myeloid malignancies.
https://doi.org/10.1182/blood.2022019304
Allogeneic transplantation for idiopathic severe aplastic anemia: analysis from the SAAWP of the EBMT
Survival after allogeneic hematopoietic stem cell transplantation (allo-HSCT) for severe idiopathic aplastic anemia (SAA) has improved, but assessing outcomes beyond survival is important. The study analyzed graft-versus-host disease and relapse/rejection-free survival (GRFS) in 479 SAA patients who underwent allo-HSCT. In the upfront cohort, where allo-HSCT was done initially, 5-year GRFS was 77%. Late allo-HSCT (>6 months after SAA diagnosis) increased the risk of death as the cause of GRFS failure. In the rel/ref cohort, where allo-HSCT was for relapsed or refractory SAA, 5-year GRFS was 61%, with age being a significant factor for increased risk of death, acute GvHD, and chronic GvHD as the cause of GRFS failure. The study suggests that early allo-HSCT is beneficial in younger patients with a matched related donor, while in salvage allo-HSCT, especially in older patients, outcomes are less favorable.
https://doi.org/10.3324/haematol.2022.281876
Deciphering treatment patterns in non-severe/moderate aplastic anemia: an international observational study
In a comprehensive study of 259 patients with non-severe aplastic anemia, various treatment strategies were employed, including cyclosporine (CyA) alone or in combination with anti-thymocyte globulin (ATG) or eltrombopag, eltrombopag alone, and others. Similar overall response rates were observed across different strategies, with notable trilineage response achieved in 39% of patients, particularly with CyA plus eltrombopag. Progression to myeloid neoplasms occurred in 8% of cases, unrelated to mutational status. Predictors of survival included age, gender, lactate dehydrogenase levels, transfusion needs, and somatic mutations, while factors like higher neutrophils, paroxysmal nocturnal hemoglobinuria clones, and trilineage response were associated with better survival. Age and trilineage response at 6 months were confirmed as significant factors in multivariable analysis.
https://doi.org/10.1038/s41375-023-02047-z
Addition of eltrombopag to immunosuppressive therapy for adults with severe aplastic anemia- Japanese Experience
In a multicenter retrospective cohort study comparing outcomes in patients receiving immunosuppressive therapy (IST) with or without eltrombopag for severe aplastic anemia, 82 patients who received front-line therapy from January 2014 to August 2021 were included. Overall response rates at 6 months did not significantly differ for patients receiving eltrombopag versus IST alone (58% vs. 65%, p = 0.56). However, complete response rates at 6 and 12 months were over two times higher in the eltrombopag arm (29% vs. 12%, p = 0.06 and 48% vs. 18%, p = 0.005). Rates of hepatotoxicity were similar across both arms, and eltrombopag addition did not impact overall survival or disease-free survival significantly. The study suggests that eltrombopag may offer deeper responses with similar toxicity rates to IST alone in real-world settings.
https://doi.org/10.1007/s12185-023-03670-3
Androgens in bone marrow failure disorders
This retrospective analysis examined the usage, effectiveness, and toxicity of androgens in bone marrow failure (BMF) syndromes, utilizing data from 274 patients across 82 European Society for Blood and Marrow Transplantation (EBMT) affiliated centers. The cohort comprised 193 patients with acquired BMF and 81 with inherited BMF, with median ages of 32 years and 8 years, respectively. Androgen treatment resulted in complete and partial remission rates at 3 months of 6% and 29% in acquired BMF and 8% and 29% in inherited BMF. Overall survival and failure-free survival (FFS) at 5 years were 63% and 23% in acquired BMF and 78% and 14% in inherited BMF
https://doi.org/10.3324/haematol.2023.282935
Granulocyte transfusions in severe aplastic anemia
This study evaluated granulocyte transfusions (GT) in 28 severe aplastic anemia (SAA) patients, finding a 50% overall survival (OS) from initial GT over a median follow-up of 551 days. Sixty-four percent survived to hospital discharge, and those with improved or stable infections at 30 days had significantly better OS. Eighty-six percent awaiting hematopoietic stem cell transplant (HSCT) underwent transplantation, with 62% surviving to post-HSCT discharge. Alloimmunization occurred in 32% of patients, indicating that while OS remains poor, GT can help bridge patients to HSCT.
https://doi.org/10.3324/haematol.2023.283826
Alternative donor transplantation for severe aplastic anemia
This study compared outcomes of stem cell transplantation (SCT) in severe aplastic anemia (SAA) patients using matched unrelated donors (MUD), mismatched unrelated donors (MMUD), and haploidentical donors (Haplo) from 2012 to 2021. MUD had the best outcomes with lower risks of graft failure, acute graft-versus-host disease (GVHD), and nonrelapse mortality. The 3-year overall survival rates were 81% for MUD, 74% for MMUD, and 63% for Haplo. MUDs are the preferred alternative donor for SAA patients without a matched sibling donor, while the choice between MMUD and Haplo requires further investigation.
https://doi.org/10.1182/blood.2024024173
Roxadustat in the treatment of aplastic anemia patients with inadequate erythroid responses
Roxadustat is a HIF prolyl-hydroxylase inhibitor that increases endogenous production of erythropoietin. This pilot study evaluated the efficacy and safety of roxadustat in 14 aplastic anemia (AA) patients with inadequate erythroid response after immunosuppressive therapy. After 8 weeks, 64.3% showed a significant hemoglobin rise, with 14.3% reaching normal levels. By the last follow-up, 71.4% responded, and 28.6% had normal hemoglobin levels. Roxadustat was generally well tolerated, with mild adverse effects in 28.6% of patients, showing promise for improving hemoglobin levels in AA patients.
https://doi.org/10.1007/s00277-024-05799-5
Comparison of efficacy of eltrombopag combined with immunosuppression in the treatment of aplastic anemia
This study compared the efficacy and safety of immunosuppressive therapy (IST) with or without eltrombopag for treating severe aplastic anemia (SAA) and very severe aplastic anemia (VSAA) in 371 Chinese patients. The combination of eltrombopag and IST significantly improved the overall response and complete response rates in SAA at 3 and 6 months. In VSAA, the addition of eltrombopag improved the 6-month complete response rate and 3-year overall survival but showed no additional efficacy beyond these measures. Liver injury increased with eltrombopag, but other toxicities were comparable.
https://doi.org/10.1007/s00277-024-05910-w
Addition of ruxolitinib to standard graft-versus-host disease prophylaxis for allogeneic stem cell transplantation in aplastic anemia patients
In this study, 35 aplastic anemia (AA) patients underwent allogeneic hematopoietic stem cell transplantation (allo-HSCT) with the addition of ruxolitinib to standard GVHD prophylaxis. Results indicated no impact on engraftment or graft function but showed enhanced CD4+ Treg recovery and significantly reduced bacterial/fungal infections. The ruxolitinib group experienced a lower incidence of moderate-to-severe acute GVHD (17.1% vs 48.6%) and had trends toward reduced chronic GVHD, higher GVHD and failure-free survival (85.7% vs 68.6%), and lower treatment-related mortality (2.9% vs 14.3%). Overall, peri-transplant ruxolitinib addition appears safe and effective in reducing GVHD with improved allo-HSCT outcomes in AA patients.
https://doi.org/10.1038/s41409-024-02266-7
First-line treatment of severe aplastic anemia: immunosuppressive therapy plus eltrombopag versus haploidentical hematopoietic stem cell transplantation
This study prospectively compared haploidentical HSCT (Haplo-HSCT) and IST with eltrombopag (IST + EPAG) as first-line therapies for severe aplastic anemia (SAA). Results showed that Haplo-HSCT led to faster transfusion independence, higher rates of complete blood count recovery (86.3% vs. 24.1%), and improved quality of life scores. Three-year overall survival (OS) was higher for IST + EPAG in patients under 40, but Haplo-HSCT had a better failure-free survival (FFS) across all ages. For very severe SAA (vSAA) and patients aged ≥40, Haplo-HSCT was more advantageous, especially in terms of FFS and quality of life.
https://doi.org/10.1038/s41409-024-02377-1
An Initiative of
Veenadhare Edutech Private Limited
1299, 2nd Floor, Shanta Nivas,
Beside Hotel Swan Inn, Off J.M.Road, Shivajinagar
Pune - 411005
Maharashtra – India

howitreat.in
CIN: U85190PN2022PTC210569
Email: admin@howitreat.in
Disclaimer: Information provided on this website is only for medical education purposes and not intended as medical advice. Although authors have made every effort to provide up-to-date information, the recommendations should not be considered standard of care. Responsibility for patient care resides with the doctors on the basis of their professional license, experience, and knowledge of the individual patient. For full prescribing information, including indications, contraindications, warnings, precautions, and adverse effects, please refer to the approved product label. Neither the authors nor publisher shall be liable or responsible for any loss or adverse effects allegedly arising from any information or suggestion on this website. This website is written for use of healthcare professionals only; hence person other than healthcare workers is advised to refrain from reading the content of this website.