
howitreat.in
A user-friendly, frequently updated reference guide that aligns with international guidelines and protocols.
Histiocytic/Dendritic cell neoplasms
Histiocytic/Dendritic cell neoplasms
- They are tumors arising from cells of mononuclear macrophage system (monocytes, macrophages and dendritic cells)
- Mononuclear macrophage system is a part of innate immunity and it is involved in detection and phagocytosis of pathogens.
- Tumors include:
- Neoplasms of conventional dendritic cells:
- Langerhans cell histiocytosis
- Langerhans cell sarcoma
- Interdigitating dendritic cell sarcoma
- Indeterminate dendritic cell tumour
- Neoplasms of histiocyte/ macrophages:
- Erdheim-Chester disease
- Rosai-Dorfman disease
- Juvenile xanthogranuloma
- ALK-positive histiocytosis
- Histiocytic sarcoma
- Neoplasms of plasmacytoid dendritic cells:
- Mature plasmacytoid dendritic cell proliferation associated with myeloid neoplasm
- Blastic plasmacytoid dendritic cell neoplasm
- Neoplasms of conventional dendritic cells:
- Most of these neoplasms occur de-novo, but some may be seen in association with lymphoma/ leukemia
Conventional dendritic cell neoplasms
Langerhan Cell Histiocytosis
- It is a clonal neoplasm of myeloid dendritic cells expressing a Langerhans cell phenotype (Positive for CD1a and CD207).
- Commonly affects skin and bones.
- Earlier Langerhans cell histiocytosis was classified into 3 categories
- Letterer-Siwe disease- Presents as cutaneous lesions resembling seborrheic eruptions
- Eosinophilic granuloma- Along with Langerhans cells there is presence of eosinophils, plasma cells and lymphocytes
- Hand Schuller Christian disease – Triad of
- Calverial bone defects (eruption over scalp and ear canal)
- Diabetes insipidus (Due to involvement of posterior pituitary stalk)
- Exophthalmos
Epidemiology:
- 5 cases/ million population/ year
- Common in childhood
- M:F= 3.7:1
- Common in Europeans
Etiology:
- Neonatal infection
- Solvent exposure in parents
- Lack of childhood vaccination
- LCH of lung – smoking
Pathogenesis:
- Tumor cells are derived from bone marrow myeloid dendritic cells
- Langerhans cell maturation is prevented by TGF beta and IL-10.
- Mutation of MAPK pathway (Ex: BRAF, MAP2K1 etc) is often found, which leads to activation of ERK pathway.
Classification:
- Single system (One organ or one system is involved):
- Bone (Single bone/ multifocal)
- Skin
- Lymph node
- Lungs
- Hypothalamic-pituitary/ CNS
- Others: Thyroid, thymus etc
- Multisystem
- Two or more organs are involved
Clinical Features:
- Constitutional symptoms: Fever, sweating, weight loss, diarrhea, edema, dyspnea
- Bone marrow involvement: Cytopenias
- Skin
- Lesions look similar to seborrheic dermatitis of scalp (Mistaken for prolonged cradle cap in infants)
- Skin flexures are commonly affected, but lesions can be generalized.
- Sometimes deep subcutaneous purplish papules are seen (Hashimoto-Pritzker disease)
- Sometimes ulcerative lesions behind the ears, involving scalp, skin of genitals or perineal region may be seen.
- Oral cavity
- Gingival hypertrophy
- Ulcers of soft and hard palate, buccal mucosa, tongue or lip
- Premature eruption of baby tooth. Erosion of lamina dura leading to "floating tooth appearance" in dental X ray
- Bone lesions
- Lytic lesions in skull- Asymptomatic or painful
- Any bone may be affected
- Spine lesions are more common in cervical region
- Bone pain, back pain, muscle pain, joint pain, osseous mass
- Ear discharge- Copious, white/ green colored
- Lymphadenopathy
- Cervical group are commonly affected
- Soft or hard matted masses with accompanying lymphedema
- Enlargement of thymus
- Hepatosplenomegaly with direct hyperbilirubinemia and hypoalbuminemia
- Lung
- Cystic and nodular lesions on CT
- Spontaneous pneumothorax
- Dyspnea, cough, hemoptysis, chest pain, diminished aeration, rales, crackles due to widespread fibrosis and destruction
- Endocrine:
- Polyurea and polydipsia- Due to involvement of posterior part of pituitary. Seen in 5-40% of patients
- Gynecomastia, decreased libido, weight changes, appetite changes, cold intolerance, constipation
- GIT
- Diarrhea, hematochezia, perianal fistula, malabsorption syndrome
- Cardiovascular
- dyspnea, orthopnea, hypertension, irregular pulse, bradycardia, cardiomegaly, murmurs
- CNS manifestations
- Neurodegenerative syndrome manifested by headaches, ataxia, dysarthria, seizures, cognitive decline, disconjugate gaze, cranial nerve palsies, ataxic or magnetic gait, sensory or motor impairment, hemiparesis, hyperreflexia, dysphagia, limb or facial weakness, sensory changes, and hearing impairment
- MRI in such cases shows hyperintensity of dentate nucleus and white matter of cerebellum on T2 weighted images)
- Atrophy of cerebellum
- Orbital involvement
- Vision loss or strabismus due to optic nerve or orbital muscle involvement
- Exophthalmos
- Genital
- Testicular mass
- Renal
- Hematuria, flank pain
- Psychiatric:
- Depression, anxiety, disinhibition, inappropriate laughing or crying, pseudobulbar affect
Investigations:
- Biopsy
- Langerhans cells: 10-15 µ in size, Oval in shape
- Nuclei:
- Grooved / folded / indented / lobulated
- Fine chromatin
- Inconspicuous nucleoli
- Thin nuclear membrane
- Cytoplasm
- Moderate, vacuolated
- Slightly eosinophilic
- No dendritic processes
- Characteristic milieu includes – eosinophils, histiocytes (sometimes multinucleated), neutrophils and small lymphocytes.
- Early- Large number of Langerhans cells, eosinophils, neutrophils
- Late- Fibrosis and foamy macrophages
- Lymph node- Involvement of sinuses with secondary infiltration of paracortical regions
- Bone marrow- Large clusters/ sheets of LCH cells with eosinophils
- Electron microscopy- Cytoplasmic Birbeck granules
- Tennis racket shape
- Zipper like appearance
- 200-400 nm with 33 nm width
- Immunophenotyping by immunohistochemistry
- Positive: CD1a (Diagnostic of LCH), S100, CD74, Vimentin, HLA-DR, Peanut agglutinin lectin, Placental alkaline phosphatase, and CD207/Langerin (Stains Birbeck granules)
- Weak positive for – CD45, CD68, Lysozyme
- Negative – B cell markers, T cell markers (with exception of CD4), CD30, myeloperoxidase, CD34, Epithelial membrane antigen, Follicular dendritic cell markers (CD21, CD35)
- Mutation analysis in the MAPK pathway
- PET scan- To detect systemic organ involvement. Active sites are PET avid.
- Urine osmolality- To find out diabetes insipidus
- Plasma FLT3 ligand, M-CSF, osteoprotegerin levels- Elevated
- Hemogram- Anemia, thrombocytopenia
- ESR- Increased
- LFT- Hypoalbuminemia, elevated enzymes and hyperbilirubinemia
Criteria for Diagnosis: Both are required
- Morphologically characteristic LCH (Histiocytes with grooved nuclei, eosinophilia)
- Immunohistochemically positive for CD1a and / or Langerin
Prognosis:
- In general, good long-term survival
- Some lesions may spontaneously regress or may repeatedly "reactivate".
- Response to initial 6 weeks therapy with weekly Vinblastine and daily prednisolone is the most important prognostic marker
- 5-year overall survival
- Responders- 79%
- Non-responders- 11%
Differential Diagnosis:
- Skin lesions: Fungal diaper rash, seborrheic scalp rash, congenital viral infection, neuroblastoma, contact dermatitis, psoriasis
- Genital lesions- Sexually transmitted diseases
- Lytic bone lesions: Neuroblastoma, rhabdomyosarcoma, Ewing sarcoma
- Ear discharge- Otitisexterna
- Collapsed vertebra- Tuberculosis, trauma, osteomyelitis
- Lung- Viral pneumonia
Pretreatment Work-up:
- History: Pain, swelling, skin rashes, otorrhea, irritability, fever, loss of appetite, diarrhoea, weight loss, growth failure, polydipsia, polyuria, dyspnoea, smoke exposure, behavioural and neurological changes
- Examination
- LN:
- Spleen:
- Liver:
- skin and scalp rashes:
- ear discharge:
- orbital abnormalities:
- gum and palatal lesions, dentition:
- soft tissue swelling:
- lesions on the genital and anal mucosa:
- WHO P. S.
- BSA
- IHC (CD1a, S100, Langerin, cyclin D1 and BRAF V600E)
- Molecular testing: BRAF V600E / NGS study including MAPK and related pathway mutations on tissue
- Grade
- Whole body PET/ CT including distal extremities (vertex to toes)
- BMA and Bx (If cytopenia +) –To rule out HLH/ myeloid neoplasm
- Liver biopsy (If there confusion b/w LCH and sclerosing cholangitis)
- CT Chest/ BAL for CD1a/ Lung Biopsy (If symptomatic/ Abnormal CXR
- MRI Head (if craniofacial bone lesions suspected/ Neurological abnormalities/ Endocrine abnormalities/ Aural discharge)
- Hearing assessment
- Endoscopy and biopsy (if there is chronic diarrhoea)
- Endocrine consultation
- MRI Spine (If spine lesions suspected)
- USG Abdomen
- Stage
- Hemoglobin
- TLC, DLC
- Platelet count
- Coagulation work up: PT: APPT: Fibrinogen:
- Urine(Early morning sample)
- Specific gravity:
- Morining usrine and serum osmolality:
- Thyroid function test
- Morning Serum cortisol, ACTH
- FSH, LH and testosterone levels
- Electrolytes: Na: K: Ca:Mg: PO4:
- LDH
- Ferritin
- HIV:
- HBsAg:
- HCV
- PFT: If pulmonary symptoms
- Chemotherapy consent after informing about disease, prognosis, cost of therapy, side effects, hygiene, food and contraception
- Fertility preservation if applicable
- PICC line insertion and Chest X ray after line insertion
- Tumor board meeting and decision
- Attach supportive care drug sheet
- Inform primary care physician
Treatment Plan:
- Special sites:
- Odontoid peg and vertebral lesions with intraspinal soft tissue extension
- Presence of lesions in these sites cause immediate risk to patient
- High Risk organs:
- Bone marrow
- Spleen
- Liver
- Lung
- CNS risk lesions (High risk of development of DI):
- Craniofacial bone involvement: Lesions in orbital, temporal, mastoid, sphenoidal, zygomatic or ethmoidal bones; maxilla/ paranasal sinuses or cranial fossa(Involvement of vault is not considered as CNS risk lesion)
- Involvement of ear, eye or oral cavity at diagnosis
Skin involvement only: Any one of following measures
- First line
- Observation with selenium or phenol-based shampoo
- Use of topical glucocorticoids
- Oral methotrexate (20mg/m2) weekly for 6 months
- May be combined with Prednisolone (20-40mg/m2 twice a day) OR 6-MP (50mg/m2- Daily) or Hydroxyurea (10mg/Kg twice a day)
- Oral thalidomide- 50-100mg HS
- Second line
- Topical nitrogen mustard- Mechlorethamine (In case of limited disease)
- Psoralen with PUVA
- Low dose RT
Single bone lesion without involvement CNS risk sites
- Local treatment (Curettage)-
- Clean margin is not necessary
- May do bone grafting if necessary
- Bisphosphonates
- Corticosteroid injections
- RT may be given in selected cases
- Should be avoided in children
- Useful especially in persistent/ recurrent lesions after curettage/ systemic therapy
- 6-12 gray (2 Gray per fraction)
Only lung involvement:
- Stop smoking
- Prednisolone as single agent
- Treatment of pulmonary hypertension and associated COPD.
- Follow up PFT every 3 months
- If there is progressive disease- Cladribine/ Systemic therapy
- Lung transplantation: Useful in patients with advanced and progressive disease
Other single-system LCH without involvement of critical organs (ie, CNS, liver, spleen)
- Observation
- Local therapy: RT/ Surgical excision
Indications for systemic therapy:
- Single system LCH with
- CNS risk lesions
- Multifocal bone lesions
- Special site lesions
- Multisystem/ Multifocal LCH
- CNS/ Pituitary involvement
- Unifocal LCH that progresses on local therapy
Systemic therapies:
- BRAF V600E mutated disease: Vemurafenib, Dabrafenib
- MAP kinase pathway mutation, or no other detectable/actionable mutation, or testing not available: Cobimetinib, Trametinib, Binimetinib, Selumetiniba
- Irrespective of mutation:
- Vinblastine, prednisolone anf 6-MP based protocol as below
- Cytarabine
- Cladribine
- Methotrexate (oral)
- Hydroxyurea
- Clofarabine
- Targeted therapies useful in certain circumstances:
- Crizotinib for ALK fusion
- Pexidartinib for CSF1R mutation
- Larotrectinib or Entrectinib or Repotrectinib for NTRK gene fusion
- Sirolimus or everolimus for PIK3CA mutation
- Selpercatinib for RET fusion
Dose adjustment for weight <10Kg
- <6 months- 50% of BSA
- 6-12 months- 75% of BSA
- >12 months- 100% of BSA
Dose adjustment:
- Start chemo when ANC >1000/cmm, PL- >1Lac/cmm (Initial chemo as well as maintenance 6MP)
- Neurotoxicity (Severe weakness , severe paresthesia, severe ilius)- Stop vinblastine temporarily and later resume at 50% of dose and then gradually increase the dose.
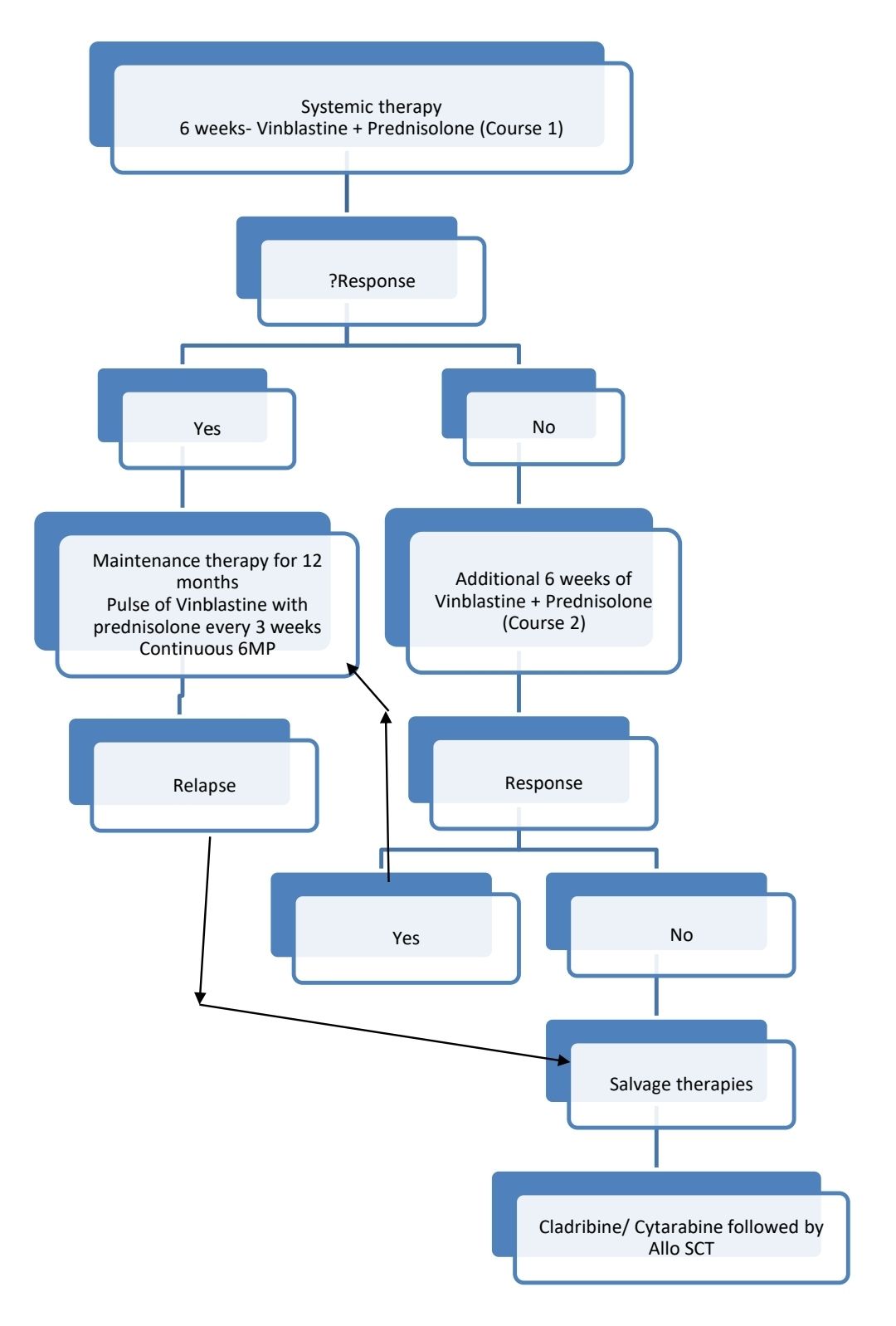
Course 1
- Tab. Prednisolone- 40mg/m2 (Max-100mg)- in 3 divided doses- From day 1 to day 28 and then taper over 3 weeks
- Inj. Vinblastine- 6mg/m2- in 100ml NS over 15min- Once a week for 6 weeks
Course 2
- Tab. Prednisolone- 40mg/m2 (Max-100mg)- in 3 divided doses- Given 3 days a week- For 6 weeks
- Inj. Vinblastine- 6mg/m2- in 100ml NS over 15min- Once a week for 6 weeks
Maintenance or continuation therapy (Each cycle of 21 days for 18 cycles):
- Tab. Prednisolone- 40mg/m2/ day- in 2-3 divided doses- From day 1 to day 5 (5 days in each cycle)
- Inj. Vinblastine- 6mg/m2- in 100ml NS over 15min- on Day 1
- Tab. 6- Mercaptopurine- 50mg/m2- OD- Continuously (Some centers avoid 6-MP in patients with low risk organ involvement)
CNS/ Pituitary mass lesion lesion- Cladribine may be used upfront
CNS neurodegenerative syndrome- Dexamethasone, cladribine, IVIg and ATRA
Patients with indications for systemic therapy, but too sick to tolerate :
- BRAF inhibitors: Vemurafenib, dabrafenib. Need to give for long time to prevent relapses.
Treatment of recurring/ refractory/ progressive disease:
- "Low risk" organ involvement
- Single bone lesion: Repeat curettage with radiotherapy.
- Repeat induction with Vinblastine weekly and daily prednisolone for 6 weeks. If there is no active disease after this, vinblastine, methotrexate and prednisolone are given every week along with daily 6MP.
- "High risk" organ involvement:
- Intensive AML like protocol which includes cladribine and cytosine arabinoside followed by stem cell transplantation
Response Criteria (by FDG-PET/CT)
- No active disease- Resolution of all signs and symptoms
- Active disease
- Regressive disease: Regression of signs and symptoms, no new lesions
- Stable disease- Persistence of signs and symptoms, no new lesions
- Progressive disease- Progressive signs and symptoms and/or new lesion
Monitoring After Treatment/ Follow-up:
- For 1 year:
- Every 6 weekly- Clinical examination, height, weight, pubertal status
- Once in 3 months: Labs: CBC, ESR, LFT, RFT
- Radiographs of bones if new lesions are suspected
- PET/CT every 3-6 months- for the first 2 years post completion of treatment
- Monitor PFTs every 3–6 months for at least 2 years for pulmonary LCH
- 2nd year to 5th year
- Every 6 months- Clinical examination, height, weight, pubertal status
- Once in 6 months: Labs: CBC, ESR, LFT, RFT
- Radiographs of bones if new lesions are suspected
- Initial positive scans- once in 2 years
Permanent consequences/ late effects:
- Diabetes insipidus transforming to panhypopituitarism
- Hearing loss
- Spinal nerve problem
- Orthopedic problems due to lesions in spine, femur, tibia or humerus
- Poor lung function in patients with diffuse pulmonary involvement
- Sclerosing cholangitis
- Dental problems
- Marrow failure
- Therapy related MDS/AML
- Higher risk of retinoblastoma, brain tumor, hepatocellular carcinoma, Ewing sarcoma
Langerhans cell sarcoma
- It is a is an aggressive malignant neoplasm of Langerhans cell
- Usually affects skin, lung, bone and soft tissue
- Microscopy: Pleomorphic histiocytes with high-grade cytology and increased mitoses
- Positive of CD1a, S100, langerin (CD207), vimentin, CD68, cyclin D1, and HLA-DR.
Interdigitating dendritic cell sarcoma:
- It is a malignant neoplasm composed of spindle to epithelioid cells showing morphologic and immunophenotypic features similar to those of interdigitating dendritic cells
- Solitary lymph node involvement is common
Indeterminate dendritic cell tumour
- It is a is a clonal proliferation of histiocytes expressing dendritic cells markers (CD1a, S-100 protein) and lacking CD207/langerin.
- Presents with skin eruptions
Histiocyte/macrophage neoplasms
Erdheim Chester disease
Introduction:
- Non Langerhans form of histiocytosis
- May be associated with Neurofibromatosis type 1 and juvenile myelomonocytic leukemia
- Commonly seen in adult men- 50-60 years
- Somatic mutation of BRAF gene (V600E) is seen in many cases
- Malignant histiocytes release proinflammatory cytokines, which attract other inflammatory cells
Clinical features
- Xanthoma like skin lesions
- Bilateral lower limb bone pain
- In case of disseminated disease- Cardiopulmonary insufficiency due to circumferential sheathing of aorta, renal failure due to retroperitoneal involvement, ataxia/DI due to CNS involvement and exophthalmos
Prognosis:
- Local lesions- Benign
- Systemic forms- Poor prognosis
Investigations:
- Biopsy- Sheets of foamy histiocytes are seen. Tumor cells are small, oval, with bland, round to oval nuclei without grooves. Cytoplasm is pink and lipid laden. Toutan giant cells are especially seen in skin lesions.
- IHC-
- Positive- Macrophage markers (CD14, CD68, CD163, PU.1, CD4, Stabilin 1, Fascin), Factor XIIIa , Cyclin D1
- Negative- CD1a, Langerin, S100, CD123, OCT2
- BRAF V600E mutation testing in biopsy samples
- ESR- Increased
- Alkaline phosphatase- Elevated
- X-Ray- Bilateral patchy osteosclerosis of metaphysis and diaphysis of femur, proximal tibia and fibula. Lytic lesions are seen in 1/3rd of cases.
- CT Chest- Diffuse interstitial infiltrate with pleural and interlobular septal thickening.
- CT abdomen- Perineural invasion, extending through the fat of the anterior and posterior pararenal spaces (Classical "Hairy kidney" appearance)
Treatment:
- May avoid treatment in completely asymptomatic patients and patients without end organ damage (Tests for diabetes insipidus must be done in all cases- Morning urine osmolality and morning serum cortisol)
- Interferon alfa-
- Used if no mutation detected even on repeat biopsy.
- Peg INF alfa- 135mcg weekly and gradually increase up to 200mcg weekly.
- Conventional INF- 3million IU (increase up to 9million IU)- 3 times a week
- Vemurafenib (480mg- BD-PO)- Approved for patients with BRAF V600F mutation. Continue treatment until disease progression/ unacceptable toxicity. Common side effects include: arthralgia, alopecia, fatigue, rash, skin papilloma etc. Avoid co-administration with CYP3a4 inhibitors.
- Other mutations (N-RAS, KRAS, ARAF, PIK3CA, MAP2K1, and ALK)- Cobimetinib (MEK inhibitor)
- Others (Avoid them as initial therapies)- Steroids, Steroids+Vinblastine, Serolimus+Prednisolone, Methotrexate, Bisphosphonates, cladribine
- Targeted therapies useful in certain circumstances:
- Crizotinib or Alectinib or Brigatinib or Ceritinib or Lorlatinib for ALK fusion
- Pexidartinib for CSF1R mutation
- Larotrectinib or Entrectinib or Repotrectinib for NTRK gene fusion
- Sirolimus or everolimus for PIK3CA mutation
- Selpercatinib for RET fusion
Rosai-Dorfman Disease
Introduction:
- It is also called as sinus histiocytosis with massive lymphadenopathy
- It is characterized by nodal or extranodal accumulation of large, S100-positive histiocytes/macrophages that commonly exhibit emperipolesis
Epidemiology:
- Common in children and young adults
- No gender/ ethnic predilection
- Annual prevalence: 1:200,000
Etiology:
- ? Immune dysregulation
- Associated with herpes virus
- Gain-of-function mutations in the MAPK/ERK pathway, including KRAS, NRAS, MAPK21, ARAF, CSF1R etc
Clinical Features:
- Lymphadenopathy-
- Massive, painless, bilateral
- Cervical commonly involved
- Fever, night sweats, malaise, weight loss
- Some have- polyarthralgia, rheumatoid arthritis, glomerulonephritis, asthma, diabetes mellitus
- Skin lesions (in 16% cases)-
- Painless maculopapular eruptions (sometimes reddish/ bluish, yellow-xanthomatous)
- Subcutaneous nodules
- Nasal and sinus involvement (Seen in 16% cases)- Airway obstruction, epistaxis, septal displacement, mass lesion infiltrating the sinuses
- Eyelid/ orbital mass with proptosis (Seen in 10% cases)
- Osteolytic lesions (Seen in 10% cases)- Irregular borders but may have sclerotic bone lesions
- Bilateral parotid/ submandibular gland enlargement
- CNS mass (Seen in <10% cases)
- Intracranial, epidural or dural
- Presents with headache, nerve palsies, syncope
- Other organ involvement (1-3% cases)- Kidney, genito-urinary, lungs, larynx, liver, tonsils, breast, GIT and heart.
Investigations:
- Lymph node biopsy
- Marked sinusoidal dilation with proliferation of foamy histiocytes within the sinuses
- Histiocytes have large, oval, vesicular nuclei and prominent nucleoli
- Rare mitosis
- Low number of eosinophils
- Plasma cells are abundant
- Emperipolesis- Lymphocytes surrounded by membranes of histiocytes
- Immunohistochemistry:
- Positive for S100, OCT2, cyclin D1, phosporylated ERK, Alpha1 Antitrypsin, CD68, CD14, CD15, lysozyme, transferrin receptor, IL-2 receptor, CD163.
- Negative- CD1a and CD207/langerin
- Targeted capture next generation sequencing of lesional tissue for mutations in RAF-RAS-MEK-ERK pathway
- Whole body PET-CT
- Hemogram
- Hemolytic anemia
- Anemia of chronic disease
- ESR- Markedly increased
- BMA and biopsy- if cytopenia
- Lumbar puncture (if brain lesions and it is inaccessible)
- S. Protein electrophoresis- Polyclonal hypergammaglobulinemia
- ALPS panel
- ANA
- Ra factor
- HLA-B27
- LDH
- MRI Brain and orbit with contrast
- PFT
- Thyroid ultrasound
- Testicular ultrasound
- RFT
- LFT- Increased liver enzymes
Prognosis:
- Most patients- Slow and steady decrease in lymph node size over months to years.
Pretreatment Work-up:
- History
- Examination
- Hemoglobin
- TLC, DLC
- Platelet count
- Peripheral smear
- LFT: Bili- T/D SGPT: SGOT:Albumin: Globulin:
- Creatinine
- Electrolytes: Na: K: Ca:Mg: PO4:
- LDH
- HIV:
- HBsAg:
- HCV:
- Whole-body FDG-PET/CT scan including distal extremities (vertex to toes)
- Serum immunoglobulins
- If autoimmune disease is suspected ALPS panel, Antinuclear antibody (ANA), antineutrophil cytoplasmic antibodies (ANCA), rheumatoid factor (RF) and HLA-B27.
- In selected cases
- CECT: chest, abdomen, and pelvis/ sinuses/ chest (high-resolution)
- MRI with and without contrast: brain/ orbit/ spine/ sella turcica ± pituitary (if diabetes insipidus suspected)
- PFTs
- Transthoracic echo (especially for suspected pulmonary RDD)
- Thyroid US
- Testicular US
Treatment:
- Many cases are self-limiting and do not require any treatment (Hence observation is adviced for asymptomatic cases)
- Surgical excision of lymph nodes, that are causing problem
- Indications for treatment
- Multiorgan involvement/ dysfunction
- Associated immune dysfunction
- Treatment:
- Glucocorticoids
- Methotrexate
- 6MP
- Cladribine
- Vinorelbine + Methotrexate
- Vinblastine
- Low dose cyclophosphamide
- Targeted therapies useful in certain circumstances:
- Crizotinib for ALK fusion
- Pexidartinib for CSF1R mutation
- Larotrectinib or Entrectinib or Repotrectinib for NTRK gene fusion
- Sirolimus or everolimus for PIK3CA mutation
- Selpercatinib for RET fusion
Response assessment:
- PET/CT after 2-3 cycles of systemic chemotherapy and at the end of treatment
Juvenile Xanthogranuloma
- It is clonal expansion of non-Langerhans cell histiocytes which have dermal macrophage phenotype
- Accounts for 0.5% of all pediatric tumors
- Most common in children <2 years old
- Lesions are confined to skin, mostly found in head, neck, upper trunk and proximal extremities
- Systemic involvement may lead to seizures, cytopenia and hepatic dysfunction
- MAPK/ERK pathway is usually involved in pathogenesis
- Microscopy: Lesion is compsed of xanthomatous histiocytes, foamy histiocytes, and Touton giant cells
- IHC:
- Positive for CD68, CD163, CD4, CD14, PU.1, factor XIIIa, fascin S-100, Cyclin D1 and OCT2.
- Negative for CD1a and Langerin
- Prognosis: Excellent
- Cutaneous lesions resolve spontaneously
- For systemic/ CNS disease, agents used include:
- Vinblastine/ Prednisolone
- Clofarabine
- Cladribine
- Cytarabine
- BRAF inhibitors
ALK-positive histiocytosis
- It is a histiocytic neoplasm lacking high-grade cytologic atypia and characterized by ALK positivity.
- Central and peripheral nervous system, bone, lung and skin are commonly involved
- Types:
- Multisystem with hematopoietic involvement (Liver/ spleen/ marrow)
- Multisystem without hematopoietic involvement
- Single system
- Usually seen in infants
- Multisystem disease presents with hepatosplenomegaly, anemia and thrombocytopenia
- Single system disease usually affects CNS
- Microscopy: Histiocytes in various forms: large oval (“epithelioid”) cells, foamy cells and spindle cells
- IHC:
- Positive: ALK, CD68, CD163, CD14, CD4 and lysozyme
- Variable: S100 protein, cyclin D1 or OCT2
- Negative: CD30, CD1a and langerin
- Treatment:
- Single system involvement: Surgical resection
- Multisystem disease/ unresectable tumors: Systemic chemotherapy/ ALK inhibitors
Histiocytic Sarcoma
- Commonly seen in lymph nodes, gastrointestinal tract, spleen, soft tissue, skin, and CNS.
- Mutations involving MAPK pathway are commonly seen.
- Often associated with other lymphoid neoplasms
- Tumor cells are positive for CD163, CD68, and lysozyme. Negative for CD1a, langerin (CD207), CD21, CD35
Plasmacytoid dendritic cell neoplasms
Mature plasmacytoid dendritic cell proliferation associated with myeloid neoplasm
- It is clonal proliferation of plasmacytoid dendritic cells with low grade morphology identified in the context of a defined myeloid neoplasm such as CMML, AML, MDS etc
- Clinical features: Lesions in skin: rash, macules, papules, and, rarely, nodular lesions
- Immunophenotype:
- Positive for CD123, TCF4, CD2AP, SPIB, CD303, CD304 and MX1.
- Negative for CD303 and TCL1
- Aberrant expression of CD34, CD56, and TdT
Blastic plasmacytoid dendritic cell neoplasm
Introduction:
- Represents <1% of acute leukemias
- Median age- 65-67 years
- 10-20% patients have other hematological malignancy such as MDS or AML or CMML.
Etiology:
- TET2 mutation is seen in 80% cases
- 12p13/ETV6 deletions are seen in most of cases
Clinical features:
- Presents with skin lesions- Solitary/ multiple, nodules/ plaques
- Regional lymphadenopathy
- Cytopenia in later stages
Investigations:
- Biopsy- Monotonous infiltration of medium sized blasts
- IHC- Positive for- CD4, CD43, CD45, CD56, Plasmcytoid dendritic cell antigens (CD123, BDCA2/CD303, CD304, TCF4, TLC1, LA, INF Alfa) Aberrant markers (CD7, CD33)
- CNS involvement must be assessed at diagnosis and CNS prophylaxis with repeated IT chemotherapy must be given.
Prognosis:
- Poor
- Median overall survival: 8 to 24 months
Treatment:
- Tagraxofusp-
- CD-123 directed cytotoxin (contains IL-3 fused to truncated diphtheria toxin).
- S. Albumin must be >3.2gm/dL before starting therapy. IV Albumin has to be given if necessary.
- Need to give prophylaxis against capillary leak syndrome (H1 and H2 histamine antagonists, paracetamol, steroids) which manifests in the form of hypotension, hypoalbuminemia, edema, weight gain and hemoconcentration.
- Dose: 12mcg/kg IV over 15min- Daily- on days 1 to 5 of a 21 day cycle. Number of cycles is not currently defined.
- Tagraxofusp treatment must be followed by maintenance Tagraxofusp/ Allo SCT
- AML like chemo, followed by Allo SCT in CR1
Recent advances:
Dabrafenib and trametinib in Langerhans cell histiocytosis and other histiocytic disorders
The combination of dabrafenib and trametinib is a well-established treatment for BRAF-mutated melanoma.The study presents dabrafenib and trametinib as effective and well-tolerated treatment options for Langerhans cell histiocytosis. Out of 34 patients treated, including those with multisystem LCH and risk-organ involvement, sustained favorable responses were observed in the majority, with some patients maintaining response for over four years. The therapy was generally well-tolerated, with some patients able to discontinue treatment without recurrence.
https://doi.org/10.3324/haematol.2023.283295
Low dose cytarabine for adult patients with newly diagnosed Langerhans cell histiocytosis
A phase 2 prospective study enrolled 61 newly diagnosed adult Langerhans cell histiocytosis (LCH) patients to evaluate subcutaneous cytarabine (100 mg/m² for 5 days) over 12 cycles. The overall response rate was 93.4%, with 32.7% achieving complete response and 60.7% partial response. After a median follow-up of 30 months, 34.4% relapsed, and estimated 3-year OS and EFS were 100% and 58.5%, respectively. Multivariate analysis identified ≥3 involved organs and baseline lung involvement as poor prognostic factors for EFS. Common grade 3-4 toxicities included neutropenia (27.9%).
https://doi.org/10.1038/s41375-024-02174-1
Mixed histiocytic neoplasms: Somatic mutations and responses to targeted therapy
Histiocytic neoplasms are diverse clonal hematopoietic disorders characterized by tumorous infiltration and uncontrolled systemic inflammation. This study examined 27 patients with mixed histiocytic neoplasms (MXH), identifying new mutations such as KRAS, MAP2K2, and BICD2-BRAF fusion. The study found that targeted therapies (BRAF or MEK inhibitors) were significantly more effective in achieving complete or partial responses and preventing disease progression compared to conventional treatments.
https://doi.org/10.1111/bjh.19462
An Initiative of
Veenadhare Edutech Private Limited
1299, 2nd Floor, Shanta Nivas,
Beside Hotel Swan Inn, Off J.M.Road, Shivajinagar
Pune - 411005
Maharashtra – India

howitreat.in
CIN: U85190PN2022PTC210569
Email: admin@howitreat.in
Disclaimer: Information provided on this website is only for medical education purposes and not intended as medical advice. Although authors have made every effort to provide up-to-date information, the recommendations should not be considered standard of care. Responsibility for patient care resides with the doctors on the basis of their professional license, experience, and knowledge of the individual patient. For full prescribing information, including indications, contraindications, warnings, precautions, and adverse effects, please refer to the approved product label. Neither the authors nor publisher shall be liable or responsible for any loss or adverse effects allegedly arising from any information or suggestion on this website. This website is written for use of healthcare professionals only; hence person other than healthcare workers is advised to refrain from reading the content of this website.