
howitreat.in
A user-friendly, frequently updated reference guide that aligns with international guidelines and protocols.
Polycythemia Vera
Updated on 10.05.2025
Introduction:
- It is a MPN that arises in a clonal hematopoietic stem cell and is characterized by increased RBC production that is independent of mechanisms that normally regulate erythropoiesis.
Epidemiology:
- Most common MPN
- Incidence – 8-12 cases / 1 million population
- Mean age – 60 years
- Male predominance
Etiology: Exact cause is not known (?Radiation exposure/ environmental toxins)
- Almost all patients have activating JAK2 Kinase mutation (JAK2 p.V617F or JAK2 exon12), which leads to EPO independent proliferation of erythroid precursors.
- Additional mutations in PV include- EZH2, ASXL, DNMT3a, TET2
- Nonrandom chromosomal abnormalities such as 20q, trisomy 8/9 etc are seen
- Impaired post translation processing of thrombopoietin receptor – mpl.
- Upregulation of Bcl-XL- So erythroid precursor cells become resistant to apoptosis.
- Increased responsiveness of progenitors of RBCs to erythropoietin or insulin like growth factor 1.
Pathogenesis:
- High risk of thrombosis is due to
- Increased viscosity
- Iron deficiency- Smaller RBCs are less deformable.
- Associated thrombocytosis
- Qualitative platelet abnormalities which lead to increased thromboxane formation.
- Leukocytosis leads to increased release of proteases and free oxygen radicals. These cause endothelial damage.
- Hemorrhages: Occur due to extensive distension of blood vessels, acquired vWD and functional platelet disorder
Phases in the course of disease:
- Prodromal (Pre-polycythemic phase)- Mild increase in RBC count
- Overt polycythemic phase- Increase in red cell mass
- Post polycythemic/spent phase- BM fibrosis leading to cytopenia and extramedullary erythropoiesis
Clinical Features:
- Asymptomatic
- Due to increased viscosity of blood – Lassitude, loss of concentration, headache, dizziness, chest pain, black outs.
- Due to engorgement of mucosal blood vessels- High color of patient (dusky brick red/ plethoric face), suffused conjunctivae, retinal vein engorgement, epistaxis.
- Splenomegaly- Seen in 30-50% of cases. It is due to extramedullary hematopoiesis. Patient may have abdominal fulness/ early satiety
- Pruritus especially after warm showers (aquagenic) – Due to release of histamine from increased number of basophils.
- Erythromelalgia
- Microarteriolar occlusion with burning sensation in digits
- It causes erythema, warmth and burning type of pain in feet and hands.
- It is often associated with digital infarction (It is treated with salicylates)
- May lead to ischemic necrosis of digits followed by auto-amputation
Complications:
- Thrombosis
- Seen in 1/3rd of patients
- Risk is high is age >65 years, previous history of thrombosis and associated cardiovascular risk factors
- 50% cases involve arteries
- Thrombosis can manifest as deep vein thrombosis (Splanchnic vein thrombosis is common), myocardial infarction, stroke, mesenteric infarction and limb infarction
- Hemorrhages resulting in GI bleeding, oropharyngeal bleeding and intracerebral / subarachnoid bleeding. Occurs due vascular engorgement and acquired vWD.
- Pulmonary hypertension- Due to release of PDGF from activated platelets, unrecognized recurrent thrombosis and smooth muscle hyperplasia.
- Gout – As high cell turnover causes hyperuricemia.
- Spent / burn out phase (Post PV MF)
- 15-20% of patients develop this after 10 years of onset of disease
- It occurs because of creeping fibrosis in marrow (myelofibrosis)
- Because of this there is myeloid metaplasia in spleen which produces splenomegaly and leucoerythroblastic blood picture
- There is reduced need for phlebotomy
- Leukemic/ MDS transformation – Risk increases after treatment with radioactive phosphorous, chlorambucil or irradiation. 20% patients develop AML.
Investigations:
- Hemogram
- Polycythemic stage
- Hemoglobin level – Elevated (14-28 gm/dL)
- Red cell count - Raised
- PCV – Raised to more than 60%
- Excess/ crowding of RBCs on smear (erythrocytosis)
- Normocyticnormochromic RBCs. If there is iron deficiency, then microcytichypochromic RBCs are seen.
- WBC count – Raised to 12-20 x 109/L, Neutrophil count is raised
- Basophilia
- Occasional immature granulocyte may be seen
- Platelet count – Raised to more than 400 x 109/L
- Many giant platelets are seen
- Spent phase
- RBC Count – Normalizes initially and then reduces
- RBC show poikilocytosis with tear drop shaped cells.
- Leucoerythroblastic blood smear
- Polycythemic stage
- Bone Marrow aspiration and biopsy
- Biopsy is done to evaluate fibrosis
- Bone marrow sample can be submitted for cytogenetics.
- Documentation is panmyelosis is a major diagnostic criterion
- Polycythemic phase:
- Hypercellular marrow which is due to proliferation of erythroid, megakaryocytic and granulocytic precursor (Panmyelosis)
- Hypercellularity extends to the subcortical marrow spaces
- Erythroid is more prominent and erythropoiesis is normoblastic. Micronormoblastic maturation is noted if there is associated iron deficiency.
- Granulocytic maturation is normal
- Normal reticulin fiber network
- Stainable iron is reduced
- Reactive nodular lymphoid aggregates are seen in 20% cases
- Megakaryocytes are increased in number, but in contrast to ET, they show marked pleomorphism with higher than normal nuclear: cytoplasmic ratio. They have hypersegmented nuclei. A characteristic admixture of large and small variants is seen. Loose clustering of megakaryocytes is seen. In ET giant megakaryocytes with hyperlobated nuclei are seen which are not seen in PV.
- Spent phase
- Reticulin and collagen fibrosis of the marrow
- Hypocellular marrow
- Megakaryocytes with hyperchromatic, dysmorphic nuclei are prominent.
- Decreased erythropoiesis and granulopoiesis
- Osteosclerosis is seen in advanced stages
- Immunophenotyping: No marker is specific
- Cytogenetics
- Nothing is specific
- +8, +9, del (20q), del (13q), loss of Y and del (1Pp)
- Abnormalities chromosome 5 and 7 are seen in association with disease progression.
- S. Erythropoietin level- Decreased or normal (<20mU/mL)- It is low even after phlebotomy
- ESR – Reduced to less than 1 mm at 1st hour
- Serum urate level – Elevated to >7 mg/dL
- Neutrophil alkaline phosphatase score – Raised
- Serum B12 levels – Elevated.
- Histamine level – elevated
- Leukocyte histidinedecarboxylase level – Raised
- Blood oxygen saturation – Normal. This helps in differentiating polycythemia vera from secondary Polycythemia
- Endogenous erythroid colony formation- Present
- JAK2 and other MPN mutation panel
- Higher JAK2V617F mutant allele burden is associated with pruritus, fibrotic transformation, and venous thrombosis
- Other mutations:
- TET2 (18%), ASXL1 (15%).
- Prognostically adverse mutations (5%–10%): SRSF2, IDH2, RUNX1, and U2AF1
- Assessment of red cell mass- Expensive and requires use of radioactive elements, hence not done.
Criteria for Diagnosis: Diagnosis requires either all 3 major criteria or the first 2 major criteria plus, the minor criterion.
- Major criteria
- Elevated haemoglobin concentration (> 16.5 g/dl in men;> 16.0 g/dl in women) or Elevated haematocrit (>49% in men; >48% in women)
- Bone marrow biopsy showing age-adjusted hypercellularity with trilineage growth (panmyelosis), including prominent erythroid, granulocytic, and megakaryocytic proliferation with pleomorphic, mature megakaryocytes (differences in size). Presence of fibrosis is acceptable for diagnosis of PV.
- Presence of JAK2 V617F or JAK2 exon 12 mutation
- Minor criterion
- Subnormal serum erythropoietin level
Criteria for Post polycythemic myelofibrosis:
- Required criteria
- Documentation of a previous diagnosis of WHO defined PV
- Bone marrow fibrosis of grade 2-3 (on 0-3 scale)
- Additional criteria (2 are required)
- Anemia/ sustained loss of either phlebotomy or cytoreductive treatment requirement for erythrocytosis
- Leucoerythroblastic blood picture in peripheral smear
- Increasing/ newly palpable splenomegaly
- Development of any 2 of 3 constitutional symptoms
- >10% weight loss in 6 months
- Night sweats
- Unexplained fever >37.5 degree C
Prognosis:
- Median survival is >10 years if appropriately treated
- JAK2 p.V617F allele burden: JAK2 p.V617F variant allele frequency (VAF) ≥ 75% is associated with major cardiovascular events (3.56 fold higher risk)
- Risk factors of leukemic transformation (Over all risk- 2-14% in 10 years)
- Age >65 years
- Leukocytosis: WBC count ≥ 15000/cmm
- Abnormal karyotype: del20q, +9, +8, double abnormalities, and complex karyotypes (≥3 abnormalities involving chromosomes 1, 5, 7, and 9).
- Cytoreductive agents: Pipobroman, 32P, chlorambucil
- Mutations: SRSF2, IDH2, RUNX1, TP53, ASXL1, SRSF2; IDH1, IDH2
- Risk factors for progression to post-PV myelofibrosis include:
- Age: ≥60 years
- Bone marrow fibrosis grade >1at diagnosis
- Abnormal karyotype: del20q, +9, +8, double abnormalities, and complex karyotypes (≥3 abnormalities involving chromosomes 1, 5, 7, and 9).
- WBC count ≥ 15000/cmm
- Palpable splenomegaly
- JAK2 p.V617F allele burden: JAK2 p.V617F VAF > 50%
- Additional somatic mutations: ASXL1, IDH and SRSF2.
- Presence of additional mutations besides JAK2 p.V617F at diagnosis
- Thrombocytosis: > 5.5 lac/cmm at diagnosis
- Mutation-enhanced International Prognostic Scoring System for PV (MIPSS-PV)
- Factors considered include:
- Age > 67 y (3 points)
- Leukocyte count ≥ 15000/cmm (2 points)
- Abnormal karyotype (1 point)
- SRSF2 mutations (2 points)
- Median survival:
- High-risk (≥ 4 points)- 3.2 years
- Intermediate-risk (2-3 points)- 13.1 years
- Low-risk (0-1 points)- 24 years
- Factors considered include:
Pretreatment Work-up:
- History: H/O Thrombosis
- Cardiovascular risk factors: Hypertension/ DM2/ Active tobacco use/ hyperlipidemia
- Examination
- BMA and Bx
- Hemoglobin
- TLC, DLC
- Platelet count
- RICOF (If bleeding manifestations)
- S. EPO level
- LFT: Bili- T/D SGPT: SGOT: Albumin: Globulin:
- Creatinine
- LDH
- Cytogenetics
- PCR- JAK 2 Mutation
- Chemotherapy consent after informing about disease, prognosis, cost of therapy, side effects, hygiene, food and contraception
- Tumor board meeting and decision
- Inform primary care physician
Treatment Plan:
High risk features
- Age >60 years
- Prior history of thrombosis
- Platelet count >5,00,000/cmm
- Progressive leucocytosis
- Symptomatic progressive splenomegaly
- Associated cardiovascular risk factors
- Severe pruritus refractory to histamine antagonists
- Disease related symptoms (Ex: Weight loss, night sweats, pruritusetc)
- Disease related major bleeding
- Frequent/ persistent need for phlebotomy or intolerant to phlebotomy
Treatment of Low risk patients:
- Every 3-6 months: Monitor for new thrombosis and evaluate for indications for cytoreductive therapy (If any of these are present, treat like high risk PV)
- Manage cardiovascular risk factors: Stop smoking, control blood pressure, weight and cholesterol.
- Aspirin- Dose: 40-100mg/ day
- Phlebotomy (250-500 mL of blood every other day or twice per week) to maintain Hct
- <45% in men
- <42% in women
Treatment of High risk patients:
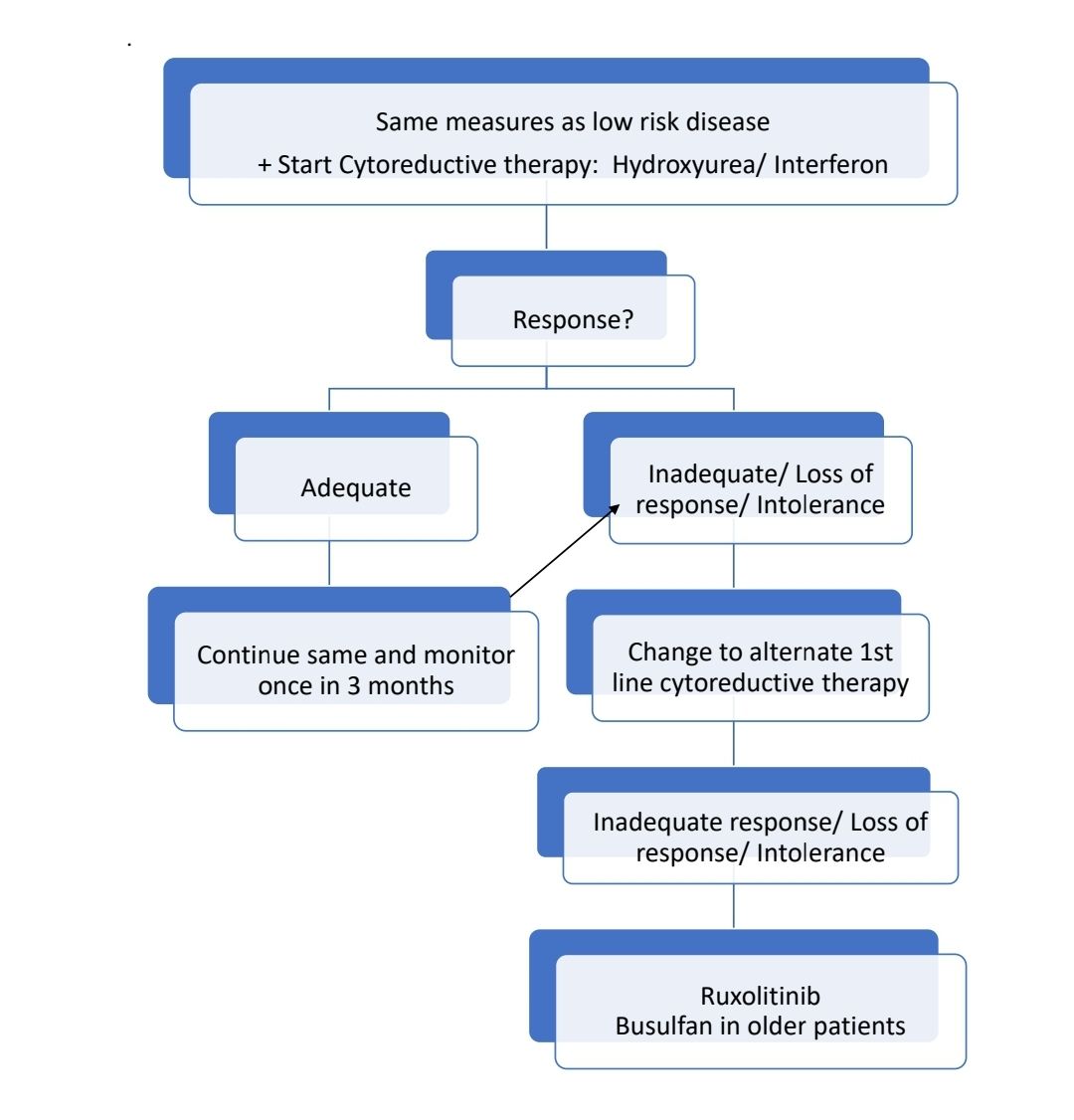
Progressed disease:
- Progression to AML: 2 options:
- Induction chemotherapy followed by Allo SCT
- Hypomethylating agents
- Progression to myelofibrosis: Treat like primary myelofibrosis
Response Criteria:
- Complete response:
- Durable (>12 weeks) resolution of disease related signs including hepatosplenomegaly, large symptom improvements
- Durable peripheral blood count remission (Hct<45%, TLC- <10000/cmm, and Platelet count <4lac/cmm)
- No thrombosis or hemorrhagic event
- No progression of disease
About Each Modality of Treatment:
- Management of cardiovascular risk factors
- Control of hypertension
- Treatment of hyperlipidemia
- Treatment of diabetes
- Stopping smoking
- Aspirin
- Given to prevent thromboembolic event
- Low dose is suggested to decrease bleeding episodes (40-100mg-OD)
- It cannot completely prevent thrombosis as WBCs are also responsible for thrombosis in PV
- Given BD if cardiovascular risk factors or leukocytosis or microvascular symptoms are present.
- Useful in alleviating microvascular symptoms.
- There is no clear recommendation on whether aspirin should be added/ stopped if anticoagulation is initiated.
- Venesection / Phlebotomy
- 400 ml of blood should be removed every 2-4 days
- Each phlebotomy decreases PCV by 3%.
- This is done to induce iron deficiency. Although iron deficiency helps to control hemoglobin levels, it increases the sense of fatigue.
- Survival is better with only phlebotomy patients compared to those on myelosuppressive therapy.
- Hydroxyurea
- 500 – 2000 mg/day – oral (Dose should be adjusted to keep platelet count less than 5 lakh/cmm and WBC count <10,000/cmm)
- Criteria for hydroxyurea intolerance/ resistance (In such situations, change to interferons/ Ruxolitinib)
- Need for frequent phlebotomy to keep Hct<0.45 after 3 months of therapy with at least dose of 2gm/day
- New thrombosis or disease related major bleeding
- Platelet count of >4,00,000/cmm or WBC count- >10,000/cmm after 3 months of therapy with at least dose of 2gm/day
- Failure to reduce massive splenomegaly to more than 50% after 3 months of therapy
- ANC <1000/cmm or Platelet count <1,00,000/cmm with lowest possible dose of hydroxyurea
- Persistence of disease related symptoms Ex: Pruritus, night sweats, fatigue
- Presence of leg ulcers or other unacceptable side effects.
- Interferon alfa-
- 3 – 5 million Units – 3 times per week – SC or Pegylated INF alpha-45-180 microgm once in 2 weeks SC.
- Preferably should be used in patients aged <40 years, pregnant females and those with intolerance to hydroxyurea
- Can cause flu like symptoms, mood changes, optic changes, emergence of autoimmunity and neuropathy.
- It can lead to complete hematological remission, reduce splenomegaly, prevent thrombosis and restoration of normal hematopoiesis.
- Usually treatment is continuous, but it can be stopped for prolonged periods of time.
- Alternates include: (They require less frequent administration and are well tolerated)
- Peg-Interferon- alpha-2a- starting dose 45 μg weekly SC injection and titrated to a maximum of 180 mcg
- Ropeginterferon alfa-2b- 100 μg subcutaneous injection every 2 weeks
- Busulfan-
- 2-4mg/day PO for 4-8 weeks and then stop once platelets <2lac/cmm or WBC <3000/cmm.
- Counts remain in normal range for months to years.
- Repeat if necessary (use dose of 2mg/day when treatment is repeated)
- Other cytoreductive agents such as Chlorambucil, Pipobroman and Radioactive phosphorous are not used now because of high risk of development of leukemia.
- Radioactive phosphorous
- 111 – 185 mBq-IV-in isotonic solution of Na2 PO4
- Indication – Old age patients with life expectancy <10 years
- Advantages- Easy administration, simple follow up and predictable effects
- Disadvantages: Leukemia in long term survivors
- Ruxolitinib
- Does not decrease malignant clone/ disease progression.
- Useful in case of symptomatic splenomegaly/ severe pruritus which fails to respond to routine cytoreductive therapy.
- Dose- 10mg- 20mg- BD. Adjust the dose based on tolerance and response.
Supportive Care:
- Gout- Allopurinol
- Anagrelide
- To prevent platelet aggregation and reduce platelet count
- Quinazoline derivative
- 1-2 mg/day in 4 divided doses.
- Used only if platelet control is inadequate with hydroxyurea.
- Side effects- Palpitation, tachycardia, nausea, diarrhea, fluid retention, headache
- Thrombosis
- Initial therapy as per ACCP guidelines
- No clear data on duration of anticoagulation.
- If there is high risk of recurrence or if thrombotic event was life threatening, better to continue anticoagulation lifelong.
- Hemorrhage:
- Transfuse platelets as platelets in PV have abnormal function
- Role of tranexamic acid is controversial
- Pruritus
- Avoid precipitating factors:
- Extremes of temperature in environment/ water used for bathing
- Adding laundry starch to bath water
- Drying the skin by patting rather than rubbing
- Moisturizing cream
- Antihistaminics (both H1 and H2) with Serotonin reuptake inhibitors
- Gabapentine/ pregabalin/tricyclic antidepressants-SSRI/ thalidomide
- PUVA therapy is useful in some cases
- Start cytoreductive therapy if not started
- Iron replacement with extreme caution
- Ruxolitinib- If above measures fail
- Avoid precipitating factors:
Special Situations:
- Surgery in PV patients
- Avoid elective surgeries in poorly controlled PV patients, as 75% of patients have hemorrhagic/ thrombotic complications. VTE risk is 5 times high even when thromboprophylaxis is given.
- Platelet count and Hct must be controlled 2 months prior to surgery, if possible.
- Use VTE prophylaxis after surgery
- PV with pregnancy
- Higher incidence of premature births, preeclampsia and hemorrhage.
- Phlebotomies should be done to maintain Hb/Hct within normal range for particular gestational age. Targets: 1st trimester- 0.31-0.41, 2nd trimester- 0.30-0.38, 3rd trimester- 0.28- 0.39
- Low risk patients should receive aspirin. Discontinue Aspirin 5 days prior to delivery.
- Patients with previous history of thrombosis, previous history of hemorrhage, previous pregnancy complications (Pre-eclampsia, >3 first trimester pregnancy losses, >1 third trimester pregnancy losses, low birth weight, intrauterine fetal death), extreme thrombocytosis, diabetes mellitus/ hypertension requiring pharmacological treatment, must receive dual anticoagulation with aspirin and LMWH (Ex: Enoxaparin 40mg/day) and also interferon for cytoreduction. Hold enoxaparin 1 day prior to delivery and restart once adequate hemostasis is achieved. Anticoagulation should be continued 6 weeks post-partum.
- Avoid iron supplementation.
- If cytoreduction is required INF-alpha is the drug of choice.
- Use routine thromboprophylaxis after delivery (LMWH) along with graded elastic stalking for all patients.
- Dehydration should be avoided during labor and 3rd stage of labor should be actively managed.
Figures:
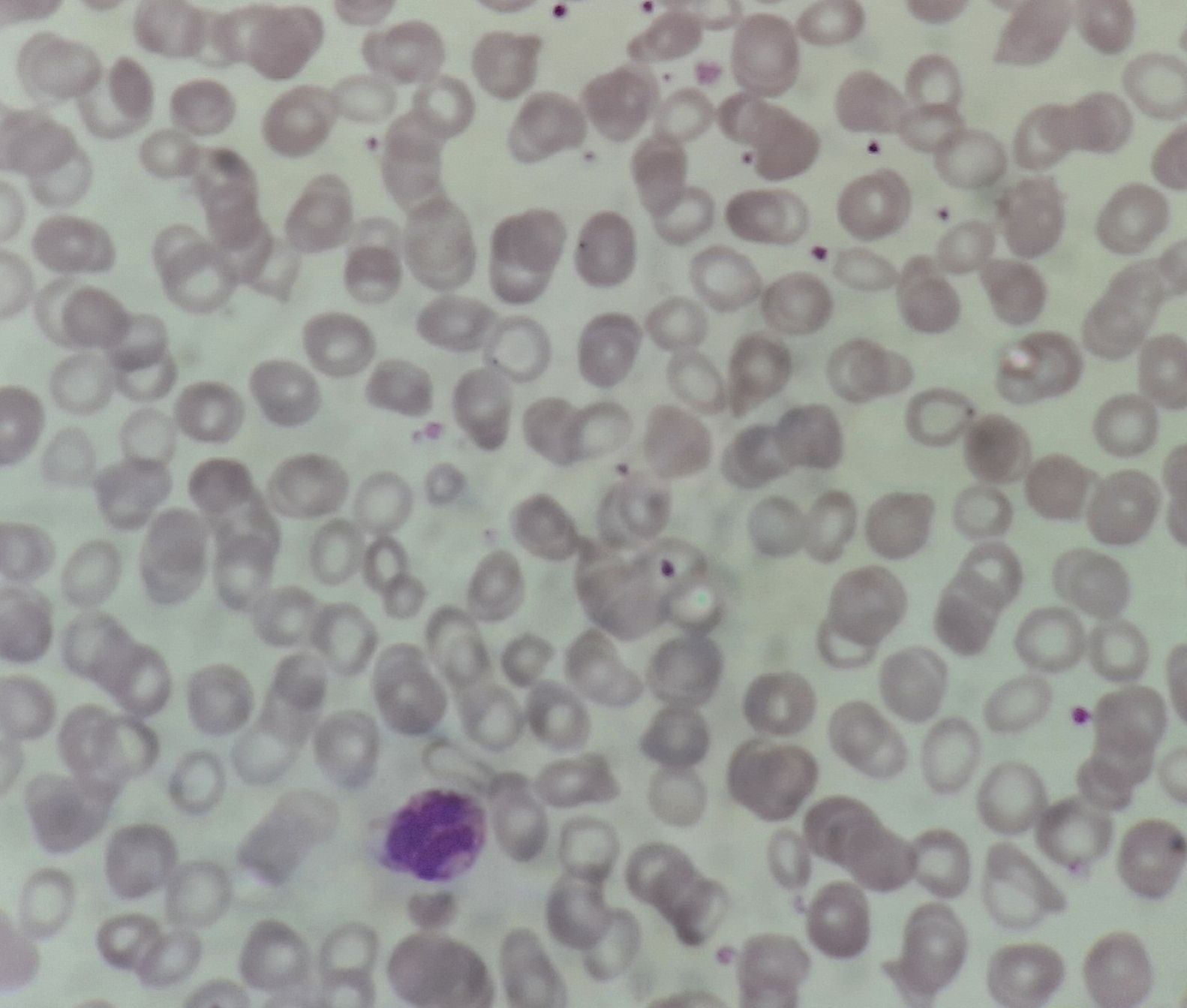
Figure 2.4.1 - Polycythemia vera- Polycythemic phase- Peripheral smear
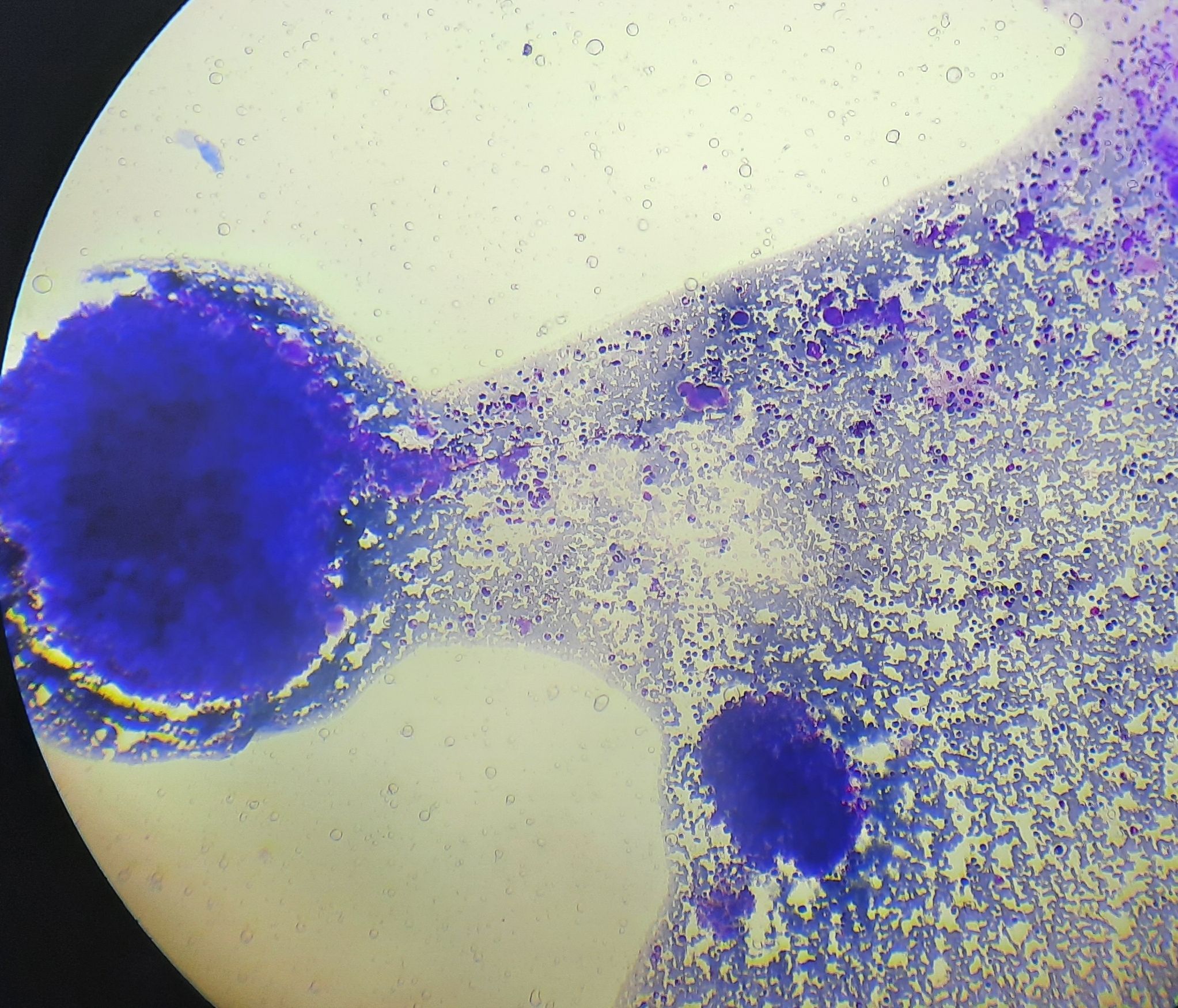
Figure 2.4.2 - Polycythemia vera- Polycythemic phase- Bone marrow aspirate
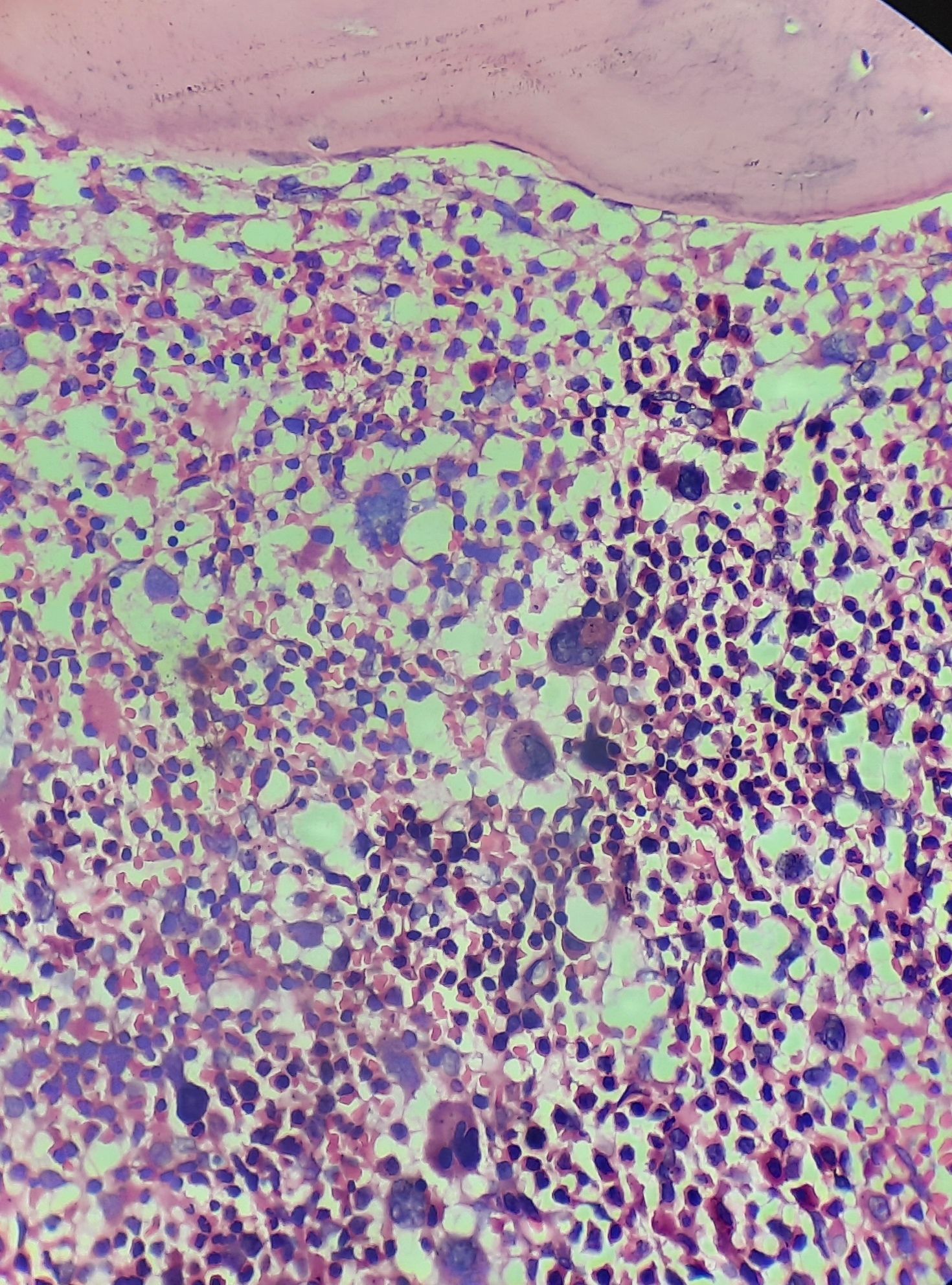
Figure 2.4.3 - Polycythemia vera- Polycythemic phase- Bone marrow biopsy
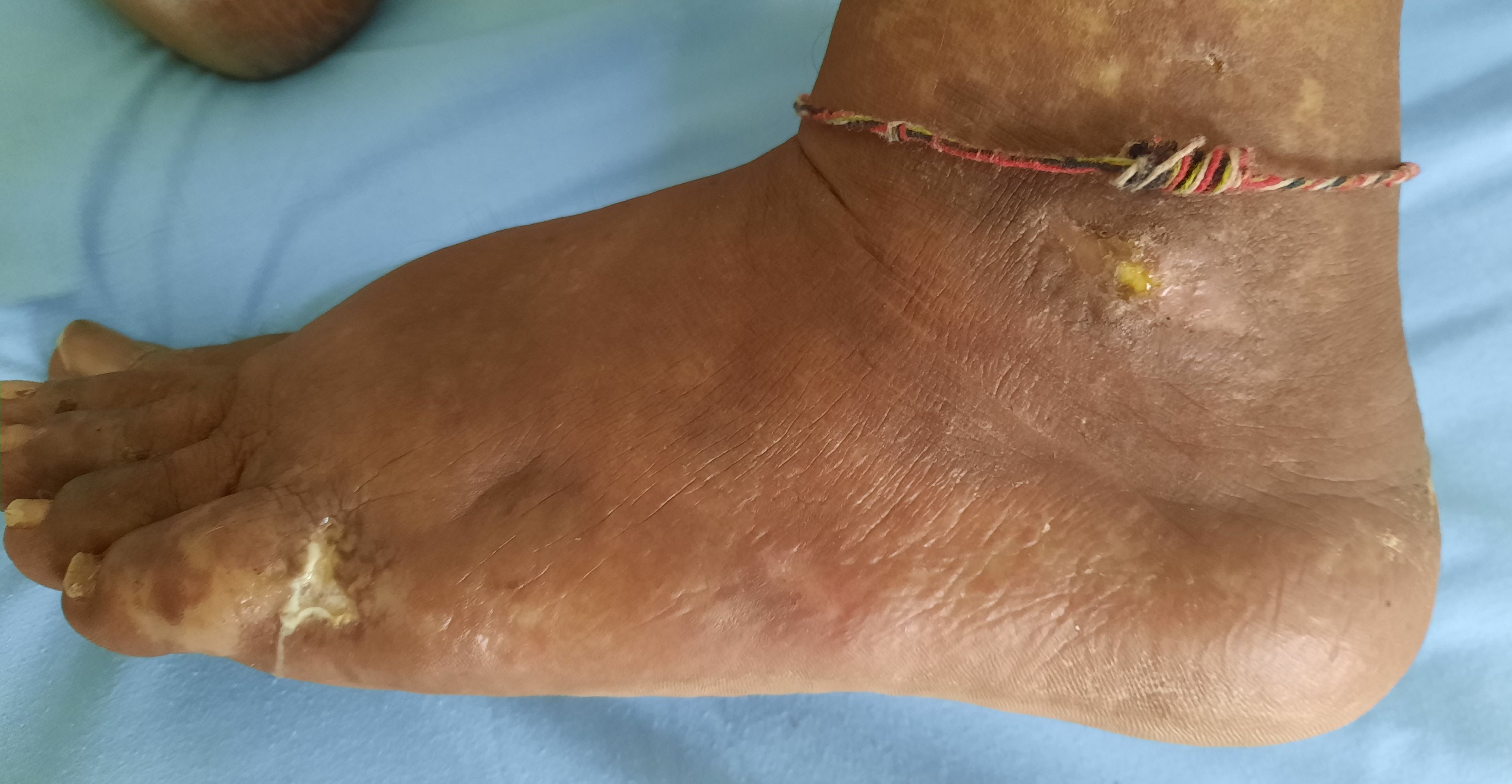
Figure 2.4.5 - Hydroxyurea induced leg ulcers
Recent advances:
Interferon-α in management of polycythemia vera and essential thrombocythemia
In a recent study by John Mascarenhas et al, role of pegylated INF alfa was evaluated in high risk PV and ET patients. Complete response rate at 12 months was 37% in HU group, while it was 35% in interferon group. PEG-INF led to a greater reduction in JAK2V617F at 24 months. Thrombotic events and disease progression were infrequent in both arms.
doi.org/10.1182/blood.2021012743
Carbon monoxide rebreathing method is a reliable test to evaluate the red cell mass in polycythaemia
The study addresses the challenge of diagnosing polycythaemia using current methods, which rely on haemoglobin and haematocrit levels as surrogate markers. While isotopic measurement is the gold standard for determining red blood cell mass (RCM), the optimized carbon monoxide re-breathing method (oCOR) offers a potentially reliable alternative. A prospective bicentric study compared RCM values obtained by oCOR and isotopic measurement in 42 patients suspected of polycythaemia. Results showed consistency between the two methods in nearly 80% of patients, with oCOR demonstrating a sensitivity of 84% and specificity of 71%. Despite slight discrepancies, oCOR proves reliable, rapid, and cost-effective, suggesting its potential use in diagnosing and monitoring polycythaemia.
https://doi.org/10.1111/bjh.19169
Rusfertide, a Hepcidin Mimetic, for Control of Erythrocytosis in Polycythemia Vera
The REVIVE trial investigated the safety and efficacy of rusfertide, a hepcidin mimetic, in patients with phlebotomy-dependent polycythemia vera. In the dose-finding phase, rusfertide significantly reduced the number of required phlebotomies and maintained hematocrit levels below 45%. In the randomized withdrawal phase, rusfertide demonstrated superiority over placebo, with 60% of patients achieving a response compared to 17% with placebo. Patient-reported outcomes also improved with rusfertide treatment, and adverse events were generally manageable, with injection-site reactions being the most common.
https://doi.org/10.1056/NEJMoa2308809
Association between elevated white blood cell counts and thrombotic events in polycythemia vera
The REVEAL study, a prospective observational study of patients with polycythemia vera (PV), found that elevated white blood cell (WBC) counts were significantly associated with an increased risk of thrombotic events (TEs) in patients with PV, independent of hematocrit levels. Specifically, WBC counts >11 × 10^9/L were associated with a 2.35-fold increased risk of initial TE occurrence. Even in patients with hematocrit levels ≤45%, WBC counts >12 × 10^9/L remained significantly associated with TE occurrence.
https://doi.org/10.1182/blood.2023020232
Efficacy and safety outcomes in Japanese patients with low-risk polycythemia vera treated with ropeginterferon alfa-2b
This post hoc analysis evaluated ropeginterferon alfa-2b in Japanese patients with low-risk polycythemia vera (PV). By week 52, 85% achieved hematocrit control and 60% achieved complete hematologic response. The JAK2 V617F allele burden decreased from 75.8% to 53.7%, with no reported thrombosis or bleeding. Treatment-emergent adverse events occurred, but no severe events or deaths were linked to the treatment.
https://doi.org/10.1007/s12185-024-03804-1
An Initiative of
Veenadhare Edutech Private Limited
1299, 2nd Floor, Shanta Nivas,
Beside Hotel Swan Inn, Off J.M.Road, Shivajinagar
Pune - 411005
Maharashtra – India

howitreat.in
CIN: U85190PN2022PTC210569
Email: admin@howitreat.in
Disclaimer: Information provided on this website is only for medical education purposes and not intended as medical advice. Although authors have made every effort to provide up-to-date information, the recommendations should not be considered standard of care. Responsibility for patient care resides with the doctors on the basis of their professional license, experience, and knowledge of the individual patient. For full prescribing information, including indications, contraindications, warnings, precautions, and adverse effects, please refer to the approved product label. Neither the authors nor publisher shall be liable or responsible for any loss or adverse effects allegedly arising from any information or suggestion on this website. This website is written for use of healthcare professionals only; hence person other than healthcare workers is advised to refrain from reading the content of this website.