
howitreat.in
A user-friendly, frequently updated reference guide that aligns with international guidelines and protocols.
Primary Amyloidosis
Introduction:
- Amyloidosis is deposition of insoluble fibrous amyloid proteins mainly in the extracellular spaces of organs and tissues
- Amyloid means starch like. This term was used as it stains brown with iodine and turns violet upon addition of dilute sulphuric acid.
- Primary amyloidosis is also called as Light chain amyloidosis and Systemic AL amyloidosis.
- Primary amyloidosis is caused by multiple myeloma and other light chain diseases such aslymphoplasmacytic lymphoma.
- Fibrils in this condition are formed by monoclonal light chains. Higher incidence is seen with lambda chain multiple myeloma.
- 15% of multiple myeloma have associated amyloidosis
- Kidney, spleen, liver and GIT are commonly involved.
- Deposition of amyloid occurs subjacent to endothelium.
- Beta - pleated amyloid protein is can be demonstrated by X ray crystallography. Deposited amyloid is Congo red positive.
Epidemiology:
- Incidence- 0.2/1,00,000 population
- Male: Female ratio is equal
- Commonly seen after 50 years of age
Pathogenesis:
Excess of immunoglobulin light chains
↓
Partially unfolded intermediates which later undergo misfolding
↓
Cross beta structure
↓
Nucleation dependent polymerization and elongation
↓
Bind to matrix components such as glycosaminoglycans (GAGs), serum amyloid P (SAP) and collagen
↓
Deposition of amyloid + Tissue damage due to cytotoxic properties of prefibrillar amyloid oligomers
Morphology:
- Involved organs are usually enlarged with waxy appearance
- On routine H & E stain- amyloid appears as amorphous, eosinophilic, hyaline, extracellular substance.
- In kidney, deposition primarily occurs within subendothelial zones of glomeruli.
- Later there can be deposition within the mesangium and interstitial peritubular region.
- In heart, deposition occurs between the muscle fibers, leading to pressure atrophy of myocardial fibers.
- In liver, deposition occurs within the space of Disse, later causing pressure atrophy of hepatocytes.
- In spleen, deposition can be limited to splenic follicles (Sago spleen) or can be large map like deposition (Lardaceous spleen)
Clinical Features: Deposition of amyloid in different organs leads to various clinical manifestations:
- Systemic effects- Weakness, dyspnoea, weight loss,
- Heart- Intractable congestive cardiac failure, Arrythmias, low voltage QRS complexes, restrictive cardiomyopathy
- Kidney- Nephrotic range proteinuria leading edema and effusions, nephritic syndrome, chronic renal failure, renal vein thrombosis, Fanconi syndrome, Renal tubular acidosis
- Liver – Hepatomegaly, portal hypertension, intrahepaticcholestasis
- Gut- Malabsorption, obstruction, hemorrhage
- Tongue- Macroglossia (Dental indentations along the sides of tongue)
- Nerves-
- Sensory motor peripheral neuropathy- Paresthesia, numbness, pain, muscle weakness
- Autonomic neuropathy: Postural hypotension (fall in systolic BP of at least 20mmg Hg when the patient has been standing for 3-5min after lying down for minimum of 5 minutes), loss of sphincter control, erectile dysfunction, altered bladder habit, early satiety, anhydrosis etc
- Blood vessels- Increased fragility
- Carpal tunnel syndrome
- Skin- Slightly raised waxy papules, periorbitalecchymosis (Black eye/ Raccoon syndrome)
- Endocrine- Hypothyroidism, hypoadrenalism
- Bleeding tendency due to
- Acquired factor X deficiency (Factor X binds to amyloid fibrils)
- Hyperfibrinolysis
- Platelet dysfunction
- Increased fragility of blood vessels
- Localized deposition can occur in any organ
- Skin/ soft tissue thickening
- Painful seronegativearthropathy
- Vocal cord nodule
- Adrenal gland involvement
- Lymphadenopathy
- Pulmonary nodule
Investigations:
- Diagnostic biopsy sites
- Abdominal subcutaneous fat pad
- Bone marrow
- Rectum
- Kidney biopsy is avoided due to high risk of bleeding
- Staining with Congo Red stain- Appears red in color under ordinary light, but has unique apple green birefringence when observed under polarised light.
- Other special stains which are used: Methyl violet, crystal violet, thioflavin, sulfated alcian blue
- Bone marrow aspiration and biopsy: Usually normal. But may show
- Features of myeloma/ lymphoplasmacytic lymphoma
- Amyloid deposition
- S. Protein electrophoresis- M Band is generally absent
- Serum and Urine Immunofixation electrophoresis and Free light chain assay
- Mass spectroscopy based microsequencing- Useful in identifying components of fibrils.IHC also can be done.
- PCR to detect known mutations in familial cases of amyloidosis
- ECG- Reduced QRS voltages
- 2 D Echo
- Median septal thickening (If it is >15mm, then median survival is <1 year)
- Poor diastolic filling
- Normal ejection fraction and contractility
- Decreased end diastolic volume.
- Doppler Echo- Best demonstrates restriction to inflow during diastole
- Iodine 123 SAP scintigraphy: Nuclear medicine technique in which radioactive SAP is injected, which binds to amyloid site. Location of amyloidosis and quantity of amyloid can be determined with this. It cannot differentiate AL from AA amyloid. It cannot detect cardiac amyloidosis due to cardiac blood pool. Rapid clearance of SAP indicates high tumor load and poor survival.
- RFT
- LFT
- PT and APTT
- NT-ProBNP- and Troponin T
- Amyloid fibril protein identification by amino acid sequencing and mass spectrometry
Indications for diagnostic evaluation for amyloid:
- Infiltrative cardiomyopathy
- Albuminuria with/ without renal insufficiency
- Peripheral neuropathy with demyelination or axonal features
- Unexplained hepatomegaly
- Carpal tunnel syndrome
- Weight loss associated with intestinal symptoms of pseudo-obstruction/ malabsorption
- Atypical myeloma
Criteria for Diagnosis:
Essential:
- Documentation of amyloid related end organ dysfunction by clinical examination (soft tissue deposition, polyneuropathy, autonomic neuropathy), supported by abnormality on tests for organ function in blood or urine
- Monoclonal protein in serum or urine or abnormal serum free light chains
- Demonstration of amyloid deposition on tissue biopsy (abdominal fat or bone marrow or salivary gland or affected organ biopsy) by Congo red (or similar thioflavin) stain or typical fibrils on electron microscopy
Desirable:
- Typing of amyloid fibril protein by laser capture followed by mass spectrometry or immunohistochemistry/immunoelectron microscopy
- Confirmation of organ involvement by imaging (echocardiography or cardiac magnetic resonance imaging for the heart)
- Mutation of one or more genes associated with hereditary amyloidosis (especially when typing by mass spectrometry unavailable or inconclusive)
Criteria for organ involvement:
- Heart: Mean left ventricular thickness in echocardiography- >12mm/ NT-Pro-BNP- >39pmol/l / Involvement noted on cardiac MRI.
- Kidney- Non Bence Jones proteinuria of >0.5gm/24hrs
- Liver- Liven span >15cm in absence of CCF
- Nerve- Axonal sensory-motor involvement by nerve conduction study / autonomic symptoms
- Lung- Interstitial infiltrates on radiological evaluation
Prognosis:
- Overall survival in untreated cases- 2years
- 5 year survival- 20%
- If there is cardiac amyloidosis (CCF)- Median survival- 8 months
- Measure NT-ProBNP- and Troponin T
- Stage 1- None increased- Median survival- 26.4 months
- Stage 2- One is increased- Median survival- 10.5 months
- Stage 3- Both increased- Median survival- 3.5 months
- Most common cause of death- Cardiac amyloidosis
- Poor prognostic markers:
- Increased creatinine
- Hepatomegaly
- Major weight loss
- Excretion of lambda chains in urine
- Elevated beta2 microglobulin levels
- Excess amyloid load
- Circulating plasma cells
- Elevated NT-ProBNP- and Troponin T
- Uric acid- >8mg/dL
- Degree of response achieved after transplant
Differential Diagnosis (Neuropathy associated with monoclonal proteins):
- MGUS associated neuropathy
- POEMS syndrome
- Cryoglobulinemia
Pretreatment Work-up:
- History
- B-Symptoms
- Examination
- LN:
- Spleen:
- Orthostatic vitals:
- WHO P. S.
- BSA
- IHC
- BMA and Bx
- Skeletal survey/ WB Low dose CT
- Cardiac status
- Hemoglobin
- TLC, DLC
- Platelet count
- Coagulation screen: PT: APTT: Factor X:
- S. Immunofixation electrophoresis
- SPEP
- S. Free light assay
- 24hr Urinary protein
- pro-BNP
- Troponin T
- LFT: Bili- T/D SGPT: SGOT: Albumin: Globulin:
- Creatinine
- Electrolytes: Na: K: Ca: Mg: PO4:
- Uric acid:
- LDH
- Beta 2 microglobulin
- HIV:
- HBsAg:
- HCV:
- Plasma cell FISH
- ECHO/ Cardiac MRI
- Stool for occult blood
- USG abdomen
- Craniocaudal liver span
- Nerve conduction study
- Pulmonary Function Test
- Chemotherapy consent after informing about disease, prognosis, cost of therapy, side effects, hygiene, food and contraception
- Fertility preservation
- PICC line insertion and Chest X ray after line insertion
- Tumor board meeting and decision
- Attach supportive care drug sheet
- Inform primary care physician
Treatment Plan: Add Doxycycline- 100mg- BD for all patients
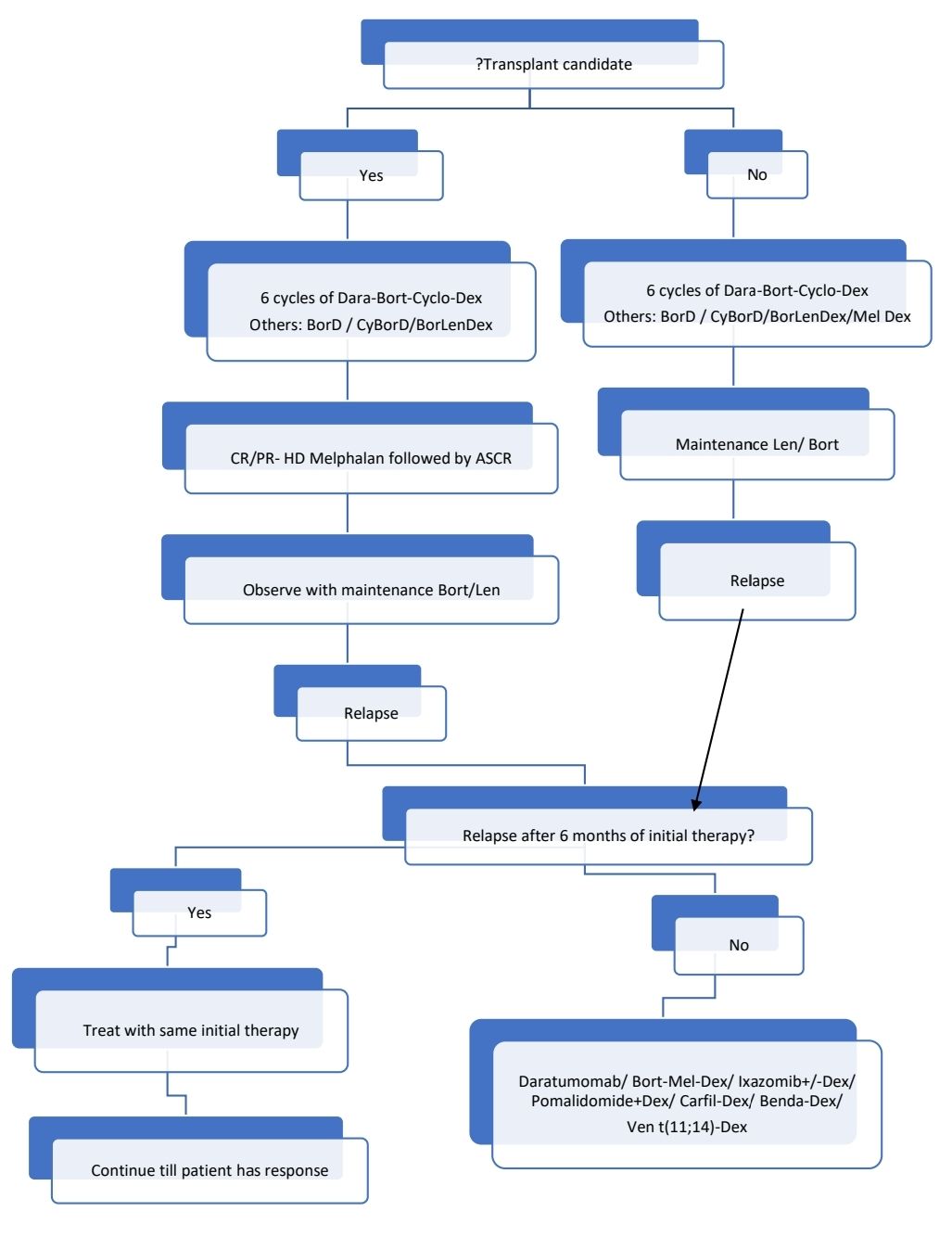
There is no standard treatment regimen. Anti-myeloma therapy must be tailored for individual patient.
Localized AL amyloidosis: If problematic, should be treated with local excision.
Response Criteria: Done by using free light chain assay done with nephelometry
- Partial response: >50% decrease in the difference between the involved and uninvolved immunoglobulin light chains, before and after therapy.
- Very good partial response: Difference between the involved and uninvolved immunoglobulin light chains is less than 4mg/dL after therapy
- Complete response: Negative immunofixation in serum and urine and normal FLC ratio.
About Each Modality of Treatment:
- Chemotherapy, similar to multiple myeloma
- Bortezomib and thalidomide should be used with caution in patients with peripheral neuropathy and cardiac involvement. Doses may be reduced in such patients.
- Autologous SCT when in CR/PR
- Melphalan- 200mg/m2
- 140mg/m2 is used if age 61-70 years, poor performance status, compensated cardiac failure, stem cell collection 2-2.5X10^6 CD34 cells/kg, Creatinine 1.5-2mg/dL
- Results are comparable to chemotherapy alone using bortezomib and thalidomide, hence avoid auto SCT in high risk patients.
- Avoid SCT in patients >70years, poor PS, significant cardiac involvement, severe orthostatic hypotension, severe renal failure.
- Consider renal transplant/ cardiac transplant prior to HDT/ASCT if there is significant renal/ cardiac damage. Following organ transplant, patients can tolerate the transplant chemotherapy better.
- Transplant related mortality- 10-15%
- Causes of death include- cardiac shock, arrythmias, GI bleeding, infections
- Maintenance therapy
- Bortezomib or Lenalidomide or Thalidomide
Supportive Care:
- Amyloid cardiomyopathy
- Sodium restriction
- Diuretics- Furosemide with spironolactone (Lasilactone)
- Use beta blockers and ACE inhibitors cautiously
- Digoxin is contraindicated, as it binds to amyloid fibrils. It can lead to severe digoxin toxicity.
- Calcium channel blockers exacerbate CCF.
- Permanent pacemaker may be needed for patients having recurrent syncope.
- Anticoagulation is useful if there is atrial stand-still
- Younger patients with advanced cardiac disease should be considered for cardiac transplantation. This procedure must be followed by chemotherapy +/- ASCT.
- Orthostatic hypotension
- Fitted waist high elastic stalking
- Midodrine (alpha 1 agonist)- Start with 2.5mg TID, and then gradually increase to 15mg TID.
- Fludrocortisone (100-200microgm/day)- Enhances fluid retention
- Continuous nor-adrenaline infusion for refractory cases
- Renal disease
- Salt restriction, diuretics
- Treatment of secondary hyperlipidemia
- Hemodialysis for end stage renal disease
- Diarrhea due to autonomic neuropathy
- Loperamide/ Opioids/ Octreotide
- Nutritional supplements
- Neuropathic pain- Gabapentin/ Duloxetine
- Lifethreatening bleeding- Rec VIIa as replacing factor X is difficult
Other Treatment Options:
- Other investigative approaches of treatment
- Destruction of deposited amyloid: Ex: Iodinated derivative of doxorubicin
- Disruption of interaction between SAP and amyloid: Ex: CPHPC
- Anti TNF alpha therapy- Eternacept- 25mg- SC- Twice weekly
- Dendritic cell based idiotype vaccination
- Anti SAP antibody treatment- Causes depletion of SAP
- Antisense oligonucleotides
Monitoring After Treatment/ Follow-up:
- SFLC assay to be done once in 3-4 months. Therapy need to be changed as soon as results start becoming abnormal.
- Every 3-4 months, following assessments must be made
- ECG, 2 D echo
- RFT
- 24hr urinary protein
- LFT
- Assessment of other organs as indicated.
Related Disorders:
- Monoclonal immunoglobulin deposition disease (Randall disease):
- Non-amyloid deposition of monoclonal immunoglobulin in tissue secondary to a plasma cell neoplasm or rarely B-cell neoplasm.
- Types:
- Light chain deposition disease
- Light and heavy chain deposition disease
- Heavy chain deposition disease:
- Most commonly kidneys are involved. Liver, heart, nerves, blood vessels are involved
- Present with Nephrotic syndrome/ chronic renal failure, arthritis and congestive cardiac failure
- Overall median survival is >5 years
- No treatment is needed for asymptomatic patients
- If symptomatic
- Lympho-plasmacytic- Treat like lymphoplasmacytic lymphoma
- Plasma cell proliferation- Treat like myeloma
- Secondary (AA) amyloidosis
- Systemic amyloidosis that occurs due to chronic inflammatory disorders such as tuberculosis, osteomyelitis, leprosy, Familial Mediterranean Fever, rheumatoid arthritis, inflammatory bowel disease, ankylosing spondylitis
- Liver produces excess of Amyloid A whenever levels of IL-1, IL-6 and TNF alpha levels are elevated.
- Treatment:
- Treatment of cause
- Eprodisate- Interferes with interaction of AA amyloid protein and glycosaminoglycans in tissues.
- Hereditory amyloidosis:
- Occurs because of production of abnormal protein
- Common types include
- Transthyretin
- Apolipoprotein
- Gelsolin
- Cystatin
- Liver transplantation is the treatment of choice for some of these conditions.
- Beta microglobulin amyloidosis
- Seen in chronically dialysed patients
- Localized amyloidosis
- Amyloidosis limited to single organ
- Different proteins are deposited in amyloidosis of different organs.
Figures:
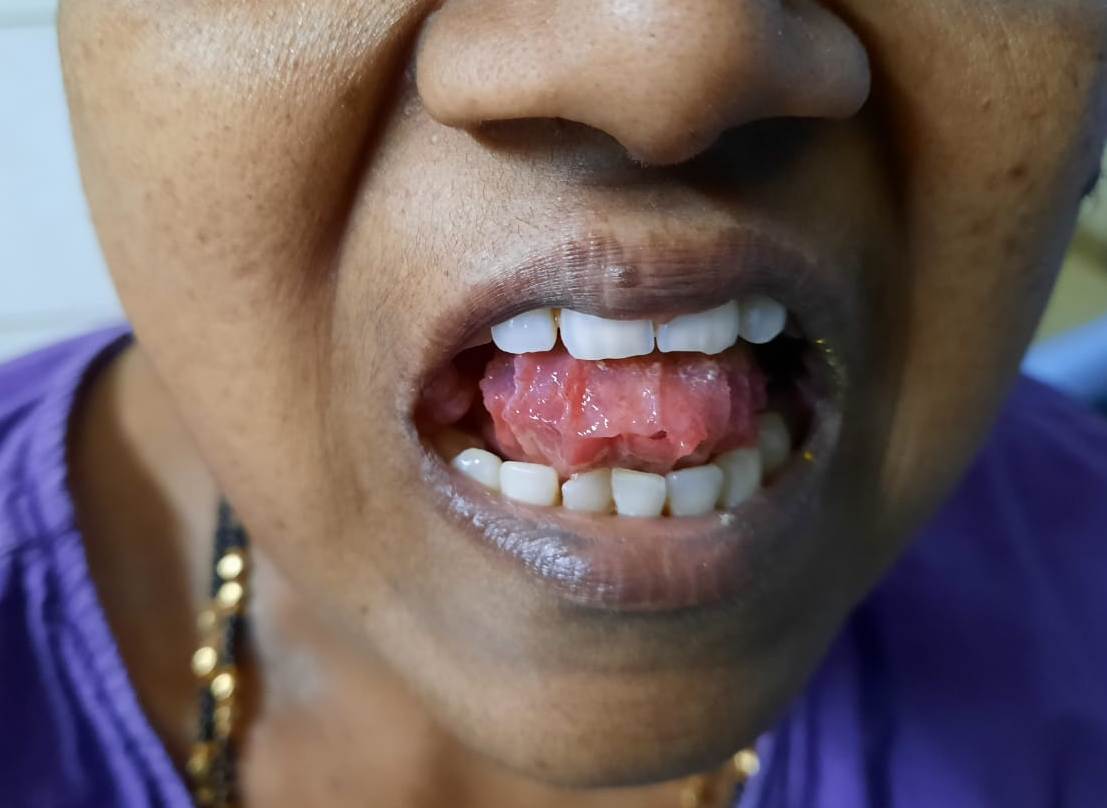
Figure 6.8.1- Primary amyloidosis- Macroglossia
Recent advances:
Novel monoclonal antibody therapy for treatment of primary amyloidosis
CAEL-101 is a novel monoclonal antibody used as passive antiamyloid immunotherapy. In a recently concluded trial this was used in patients with persistent organ dysfunction despite response to previous chemotherapy. 2/3rd of patients showed cardiac response and 20% attained renal response. The median time for response was only 3 weeks.
https://doi.org/10.1182/blood.2020009039
Primary amyloidosis is not multiple myeloma
In a recent study, published in Blood, Almeda et al have explained why primary amyloidosis and multiple myeloma are same. They studied transcriptomic atlas of plasma cells from patients with amyloidosis and multiple myeloma. They found that plasma cells of AL amyloidosis resembled secondary lymphoid organ plasma cells, where was myeloma cells resembled peripheral blood plasma cells.
https://doi.org/10.1182/blood.2020009754
Early cardiac response is possible in stage IIIb cardiac AL amyloidosis and is associated with prolonged survival
Present study evaluated the impact of early cardiac response and its depth on outcome in 249 patients with newly diagnosed stage IIIb cardiac AL amyloidosis. After a median follow-up of 52 months, 219 (84%) patients died, and median survival was 4.2 months. At 90 days, 8% patients achieved a cardiac response Cardiac response resulted in longer survival (median, 54 months).
https://doi.org/10.1182/blood.2022016348
Birtamimab plus standard of care in light-chain amyloidosis
The VITAL phase 3 clinical trial investigated birtamimab, monoclonal antibody designed to neutralize toxic light chain aggregates, in newly diagnosed patients with AL amyloidosis. The trial was terminated early after an interim analysis, as the primary composite endpoint (time to all-cause mortality or cardiac hospitalization) did not show a significant difference between the birtamimab and placebo groups. However, in a post hoc analysis of patients with the highest risk (Mayo stage IV AL amyloidosis), birtamimab demonstrated a significant improvement in survival at month 9. Treatment-related adverse events were generally similar between the two groups. A confirmatory phase 3 trial is currently enrolling patients with Mayo stage IV AL amyloidosis to further evaluate birtamimab.
https://doi.org/10.1182/blood.2022019406
Single-agent belantamab mafodotin in relapsed systemic AL amyloidosis
In a study on relapsed systemic light chain (AL) amyloidosis, belantamab mafodotin, a BCMA-directed drug–antibody conjugate, was assessed for efficacy and tolerability. Thirty-one patients, traditionally excluded from clinical trials and with a median of three prior therapy lines, were treated. The median follow-up was 12 months, with a median of five doses administered. The best hematological overall response rate was 71%, and the complete/very good partial response rate was 58%. Keratopathy was observed in 68% of patients but showed improvement in all cases.
https://doi.org/10.1111/bjh.19286
An Initiative of
Veenadhare Edutech Private Limited
1299, 2nd Floor, Shanta Nivas,
Beside Hotel Swan Inn, Off J.M.Road, Shivajinagar
Pune - 411005
Maharashtra – India

howitreat.in
CIN: U85190PN2022PTC210569
Email: admin@howitreat.in
Disclaimer: Information provided on this website is only for medical education purposes and not intended as medical advice. Although authors have made every effort to provide up-to-date information, the recommendations should not be considered standard of care. Responsibility for patient care resides with the doctors on the basis of their professional license, experience, and knowledge of the individual patient. For full prescribing information, including indications, contraindications, warnings, precautions, and adverse effects, please refer to the approved product label. Neither the authors nor publisher shall be liable or responsible for any loss or adverse effects allegedly arising from any information or suggestion on this website. This website is written for use of healthcare professionals only; hence person other than healthcare workers is advised to refrain from reading the content of this website.