
howitreat.in
A user-friendly, frequently updated reference guide that aligns with international guidelines and protocols.
Sickle Cell Anemia
Introduction:
- It is a genetic disorder characterized by presence of sickle shaped RBCs, resulting from mutation affecting hemoglobin gene.
Epidemiology:
- Prevalence is highest in Africa
- Common in northern Mediterranean countries, Saudi Arabia and central India
Etiology:
- Autosomal recessive inheritance
- It results from a point mutation that leads to substitution of polar valine for non polar glutamic acid at 6th position of A-3 helix of β- Chain
- There are several restriction fragment length polymorphisms identified around this abnormal gene. This determines the clinical manifestations in a particular population.
Pathogenesis:
Deoxygenation of HbS occurs at O2 tension of 50-60 mmHg
↓
Conformational change, which exposes a hydrophobic patch on the surface of the globin chain at the site of β6 valine
↓
Binding of this site with complementary hydrophobic site on subunit of another Hb tetramer
↓
Formation of large polymer which consist of staggered Hb tetramers that aggregate in to 21nm diameter helical fibers with one inner and 6 peripheral double strands.
- 3 Stages of polymerization
- Nucleation/lag phase (Homogeneous nucleation) – Aggregation of about 15 hemoglobin molecules into clusters
- Polymerization stage (Heterogeneous nucleation) – Polymerization into 14 strand fibers using nuclei of previous stage as starting points.
- Alignment stage – Fibers align into bundles, so cell acquires a crescent /holy leaf shape
- This crescent shape is initially reversible, but as the sickle cells loose potassium and water by Gardos pathway and potassium chloride cotransporter, they become permanently sickled. This irreversibility is due to permanent deformation of submembrane skeletal lattice.
- As these RBCs are less deformable, they are rigorously removed by spleen and liver, so life span of RBCs is reduced to 14 days which causes hemolytic anemia.
- Erythrostasis in spleen causes thrombosis and infarction which later results in autosplenectomy (Progressive shrinkage of spleen)
- Factors enhancing formation of HbS polymers upon deoxygenation
- Temperature of more than 37oC
- Higher hemoglobin concentration
- High MCHC of HbS
- Acidosis
- Hypertonicity
- Low O2 tension – Sickling starts at O2 saturation below 85% and complete at 38% O2 saturation
- 2, 3 DPG
- Dehydration (leads to increased viscosity)
- Prolonged microvascular transit time (Ex: Inflammation)
- Note: Associated HbC- Decreases polymerization
- So sickling is commonly seen in spleen, kidney, retina and bone marrow, as they have hypoxic, acidotic, hypertonic microenvironment.
- Vaso-occlusion is initiated by adhesion of young deformable red cells to the vascular endothelium, and is followed by trapping of rigid, irreversibly sickled cells
- Adhesion occurs in post capillary venules.
- Molecules involved in adhesion of RBCs to endothelium includeVCAM1, Integrins, P Selectin, Lutheral blood group antigen, Thrombospondin
- Adhesion is promoted by leucocytosis, platelet activation, and inflammatory cytokines.
- Once vaso-occlusion is resolved, there is risk of ischemia reperfusion injury.
Classification: 2 clinical types
- Sickle cell trait (heterozygous state)
- Only 40% of Hb in each RBC is HbS
- Sickling occurs only in severe hypoxia
- Full blown disease (Homozygous state)
- Most of Hb is HbS
- Children manifest the disease at 6months of age when HbF level starts declining.
- Beta gene resides in cluster of beta like genes within which there are various non-exonic polymorphic sites. Different combinations of these define discrete beta locus background haplotypes. They include:
- Senegal
- Benin
- Bantu
- Cameroon
- Arab-India
- Senegal and Arab-India subtypes are associated with high levels of HbF
- Based on clinical manifestations, there are 2 subtypes:
- Predominant hemolysis: Free hemoglobin mops nitric oxide which leads to endothelial dysfunction. They present with pulmonary hypertension, leg ulcers, priapism and stroke.
- Predominant vaso-occlusion: High hemoglobin levels lead to high viscosity of blood with subsequent vaso-occlusion. They present with acute chest syndrome, frequent painful episodes, avascular necrosis and retinopathy.
Clinical Features:
- Presents before 2 years of age, but after 6 months, when HbS predominates over HbF
- Periods of normal functioning despite chronic hemolytic anemia, punctuated by periods of vaso-occlusive painful crisis or hemolytic crisis are present.
- Other crisis which are seen are- aplastic crisis and splenic sequestration crisis.
- Pain crisis
- Bone pain and abdominal pain
- It is the most common presentation
- Precipitating factors include- Cold, dehydration, infection, stress, menses and alcohol consumption
- Generally has 4 phases-
- Prodromal phase- Low intensity pain, paresthesia, decreased red cell deformability, increase in irreversibly sickled cells
- Evolving phase- Increasing pain and worsening hematological parameters
- Established (inflammatory) phase- Steady, severe pain. increasedhemolysis, neutrophils and acute phase reactants.
- Resolution phase- All above parameters gradually revert to baseline.
- Pain has sensory, perceptual, cognitive and emotional components
- Common in chest, lower back and extremities
- Occurs due to
- Avascular ischemic necrosis commonly in femur and humerus. AVN of femoral head is seen in 50% of patients by age of 33 years.
- Vertebral body infarction and subsequent collapse leads to classical "fish mouth” appearance
- Hand- Foot syndrome
- Painful infarction of digits and ductylits
- Occurs early in infancy
- Chronic arthropathy
- Infections:
- Common organisms include: Streptococcus pneumoniae, Hemophilus influenza type B, Salmonella, E. Coli, Mycoplasma, Chlamydia, Hepatitis
- Predisposing factors in SCD are hyposplenism (autosplenectomy) and deficiency of opsonic antibodies.
- Can present with pneumonia, septicemia, meningitis, septic arthritis, osteomyelitis (Salmonella and staphylococcal), UTI
- Septicemia and meningitis caused by Strep. Pneumoniae and H. Influenzais the most common cause of death
- Ocular symptoms-
- Retinal vessel occlusion leading to
- Hemorrhage (Superficial- Salmon patch appearance, Deep-Black sun burst appearance)
- Neovascularization- Marine invertebrates/ sea fan appearance- Spontaneous regression of neovascularization occurs in 60% of cases
- Retinal detachment
- Abnormal comma shaped conjunctival vessels
- Iris atrophy
- Retinal pigmentary changes
- Orbital marrow infarction (Orbital compression syndrome)- Fever, headache, orbital swelling, visual impairment secondary to optic nerve compression.
- Retinal vessel occlusion leading to
- Renal problems
- Acidotic, hypoxic and hypertonic environment of renal medulla promotes sickling, eventually leading to ischemia
- Loss of medullary function (which results in inability to concentrate urine)
- Development of collaterals leads to disturbed counter current mechanism
- Renal papillary necrosis is seen in 1st year of life
- Bleeding is common from left kidney due to "Nut cracker" phenomenon- Longer right renal vein is compressed between aorta and superior mesenteric artery, which leads to increased venous pressure.
- Glomerulopathy- Glomerulomegaly, focal and segmental glomerulosclerosis
- Renal tubular acidosis and failure to excrete potassium
- Nephrotic syndrome
- Chronic renal failure is seen in 3rd -4th decade. Glomerularhyperfiltration and tubular secretion give falsely normal results of S.Creatinine. S.Cystatin C correlates closely with GFR.
- Causes of acute renal failure include:Hypovolemia, sepsis and hepatorenal syndrome,cardiac failure, renal vein thrombosis and rhabdomyolysis.
- Renal medullary carcinoma
- Neurological complications
- Stroke:
- Occurs in 25% of patients
- 70-80% are due to large vessel occlusion and rest are due to microvessel occlusion
- Stenosis of major vessel with formation of friable collaterals causes puffs of smoke appearance on angiography.
- Risk increases with lower baseline Hb, low HbF levels, high leukocyte count and high systolic BP
- Silent cerebral infarction occurs in young children and after 30 years of age. This leads to cognitive impairment.
- Hemorrhage
- Common between 20 to 30 years
- Most common is subarachnoid hemorrhage
- Seizures
- Unexplained coma
- Spinal cord infarction/ compression
- CNS infection
- Vestibular dysfunction and sensory-neural hearing loss
- Stroke:
- Priapism (Vascular engorgement of penis)
- It is caused by vaso-occlusion leading to obstruction to venous drainage from penis
- It has devastating psychological consequences
- It affects corpora cavernosa alone resulting in hard penis with soft glans
- It is seen in 2/3 rd of males
- Peak incidence in 2nd or 3rd decades
- Repeated episodes can lead to corporeal fibrosis and later impotence.
- Other causes of infertility include disruption of hypothalamo-pituitary- gonadal axis due to iron overload, zinc deficiency, vaso-occlusion in testicular arteries and sperm abnormalities.
- Chronic lower leg ulcers
- Occur near lateral/medial malleolus
- Seen in 2-40% cases
- Associated with hemolytic phenotype.
- They occur due to occlusion of skin vasculature by sickled cells
- These ulcers are often colonized by Ps. Aeruginosa, Staph. Aureus, streptococcus and bacteroids
- Lung problems
- Pulmonary hypertension
- Seen in 1/3rd of adult patients
- Median survival after development of pulmonary hypertension- 26 months
- Occurs due to NO scavenging by free hemoglobin and multiple thrombi in pulmonary vessels.
- Present with dizziness, fatigue, dyspnea, chest pain, syncope
- Echo- Tricuspid regurgitant jet velocity- >2.5m/sec
- Acute chest syndrome
- It is defined as new infiltrate on chest X ray and one or more of the following
- Fever
- Cough
- Chest pain
- Dyspnea
- Tachypnea
- Hypoxaemia
- It occurs due to in situ sickling within lung
- It can be triggered by
- Pain crisis
- Pulmonary infection
- Fat embolism
- Differential diagnosis include
- Pulmonary embolism
- Fluid overload
- Opiate narcosis
- Alveolar hypoventilation due to pain
- Seen in 29% of patients
- Has a mortality rate of 10% (High risk of death if age >20 years, Platelet count >2lac/cmm, multilobar lung involvement, history of cardiac disease)
- It is defined as new infiltrate on chest X ray and one or more of the following
- Chronic restrictive lung disease
- Recurrent chest infections
- Pulmonary embolism
- Pulmonary hypertension
- Hepatobiliary problems
- Pigment gall stones due to chronic hemolysis with cholecystitis in some cases
- Hepatomegaly and liver dysfunction due to
- Intrahepatic blood sequestration
- Transfusion related hepatitis
- Transfusion related iron overload
- Benign cholestasis- Causes asymptomatic hyperbilirubinemia
- Hepatic crisis- Liver infarction leading tofever, right hypochondriac pain,leucocytosis, icterus and enlarged tender liver
- Sickling crisis
- It is acute sickling leading to ischemia of various organs
- Precipitating factors include infection, fever, excess exercise, anxiety, abrupt change in temperature, hypoxia, hypertonic dyes, dehydration and acidosis
- Aplastic crisis
- Occurs when there is associated parvovirus infection
- Characterized by abrupt fall in hemoglobin and reticulocyte count
- Erythropoiesis recommences in 7-10 days
- Recurrence is rare, due to development of antibodies to Parvovirus B19
- Splenic sequestration crisis
- It is acute venous obstruction of spleen
- It occurs in 30% of patients.
- Usually seen at 6-12 months of age
- Results in rapid increase in size of spleen, massive decrease in erythrocyte mass, thrombocytopenia, hypovolemia, and shock
- ASPEN Syndrome- Association of Sickle cell disease, Priapism, Exchange transfusion, and Neurological events
- Psychosocial issues
- Depression
- Low self esteem
- Social isolation
- Poor family relationships
- Withdrawal from normal daily living
Investigations:
- Hemogram
- Hemoglobin content- Reduced to 6-10 gm/dL
- Hematocrit – 0.18 – 0.30
- Normocytic normochromic RBCs
- Moderate anisocytosis and poikilocytosis
- Irreversibly sickled cells are seen – ovoid- cigar shaped – boat shaped cells.
- They are densely hydrated cells with high specific gravity, low MCV, high MCHC and increased content of calcium
- They start appearing after 3-4 months of age when HbF starts declining
- Few target cells may be seen
- Many polychromatophilic cells and few nucleated RBCs may be seen, which indicates, increased erythropoiesis
- Signs of hyposplenism: Howell Jolly bodies, target cells, basophilic stippling and siderocytes
- WBCs – Count is elevated with shift to left neutrophilia
- Platelets – Count is elevated due to loss of splenic function.
- RDW – Elevated
- Reticulocyte count – 10-20%
- Bone marrow examination: Erythroid hyperplasia to 4-5 times
- ESR-Low- Because sickled cells prevent rouleaux formation
- Serum unconjugatedbilirubin – Raised
- Serum LDH – Raised
- Serum haptoglobin and hemopexin- Reduced.
- Sickling test – Positive- Performed by using reducing agent such as sodium metabisulphite
- Hemoglobin electrophoresis
- On cellulose acetate at pH 8.4- HbS is the major hemoglobin seen and it appears as a band midway between HbA and HbA2
- HbA levels are reduced.
- Electrophoresis on citrate agar at pH 6.2 or thin layer isoelectric focusing permits separation of HbS, HbF, Hb D and Hb G. (All these have same mobility at pH 8.4)
- Hemoglobin solubility test
- Reducing agent such as sodium dithionite is added to hemolysate.
- DeoxyHbS is insoluble and renders the solution turbid
- False positive in case of unstable hemoglobin with Heinz bodies and rare Hb variants – Ex- HbC, Hb Harlem, HbI
- Oxygen disassociation curve- Shift to right indicating decreased oxygen affinity.
- X-ray
- Rarefaction and cortical thinning
- Gross expansion of medullary space due to erythroid hyperplasia
- Molecular studies (PCR)
- To detect point mutation in globin gene sequence
- Useful especially in prenatal diagnosis
Pretreatment Work-up:
- History
- Examination
- Hemoglobin
- TLC, DLC
- Platelet count
- Peripheral smear
- Reticulocyte count
- Hemoglobin electrophoresis
- LFT: Bili- T/D SGPT: SGOT: Albumin: Globulin:
- Creatinine
- Electrolytes: Na: K: Ca: Mg: PO4:
- Uric acid:
- LDH
- HIV:
- HBsAg:
- HCV:
- Iron profile: Iron: TIBC: Ferritin
- RBC Alloantibody screen
- Renal ultrasonography and urine routine
- Chest X ray
- 2D Echo
Monitoring:
- Once in 3 months- History and physical examination, CBC with reticulocyte count
- Once in 6 months- RFT, LFT
- Once a year- Hb Electrophoresis, Urine analysis, Ophthalmoscopic examination, Transcranialdoppler (if TCD velocities are >200cm/sec, repeat after 4 weeks, if it remains high, better to give chronic red cell transfusions for primary stroke prevention)
- Once a year (start at 5 years)- PFT, Dental examination, Ophthalmoscopy, 2D Echo (For Tricuspid regurgitation velocity)
Treatment:
- All patients must receive:
- Penicillin prophylaxis
- Started at 2 months of age
- Penicillin V – Oral- Dose- <3 years- 125mg BD, >3 years- 250mg- BD Or Benzathine penicillin – IM once a month
- This can be stopped after 5 yrs of age in absence of pneumococcal sepsis or splenectomy
- Vaccination
- Pneumococcal (PPV-23)- at 2 years and then after 5 years. Some guidelines suggest repeat doses every 5 years.
- Meningococcal- 2 doses with 1 month interval, then booster every 5 years.
- H. influenza B- 3 doses with interval of 1 month
- Hepatitis – B- 3 doses- 0, 1 and 4months
- Influenza virus- Annually
- Hydroxyurea·
- It inhibits ribonucleotidereductase, leading to S phase arrest of replicating cells
- Mechanisms of action in SCD
- Stimulates production of HbF
- Increases water content & deformability of RBCs
- Decreases adherenceof RBCs to vascular endothelium
- Decreases WBC count thereby decreasing endothelial inflammation
- Increases nitric oxide release
- Increases the survival in SCD patients
- Results 50% reduction in painful episodes and acute chest syndrome.
- It has no long-term mutagenic effects
- Better to give to all patients
- Compulsory indications
- Frequent pain episodes (3 or more)
- Avascular necrosis of bone
- Acute chest syndrome
- 3 or more transfusions per year
- Sickle nephropathy
- CNS manifestations/ Abnormal transcranial Doppler velocities (170-200cm/s)
- Contraindications – Children < 4 yrs of age, pregnancy
- Dose – Start with 15mg/Kg per day & increase to 25 mg/Kg per day with target ANC- 1500-3000/cmm
- Monitor- Blood counts, renal function and hepatic function
- Folic acid- 5 mg-OD
- Crizanlizumab (P selectin inhibitor)
- Inhibits red cell adherence to endothelium
- Effective in decreasing vaso-occlusive crisis
- No major side effects
- Dose- 5mg/Kg as IV infusion over 30min at week 0, week 2, and every 4 weeks thereafter.
- L-Glutamine (Endari)
- Decreases oxidative stress
- Given to patients with frequent crisis
- Dose- 5-15gm- mixed with food- BD
- Iron chelation if chronically transfused
- Penicillin prophylaxis
- Transfusion support
- Uses of transfusions include correction of anemia and reduction in the amount of circulating HbS
- Indications for blood transfusion:
- Anemia due to hemolysis
- Aplastic crisis
- Splenic sequestration crisis
- Abnormal transcranialdoppler study
- Indications for exchange transfusion (can be done with apheresis or by simple phlebotomy to remove patient's blood followed by infusion of normal RBCs. Target HbS- 30% and hemoglobin level- 9-10gm/dL)
- Stroke
- Progressive acute chest syndrome
- Persistent priapism
- Preparation for general anesthesia
- Retinal artery occlusion
- Transfusions are not indicated in:
- Compensated anemia
- Uncomplicated acute painful crisis
- Uncomplicated pregnancy
- Avascular necrosis
- Infection
- Minor surgery without anesthesia
- Complications of transfusions
- Alloimmunization to minor blood group antigens
- Hyperviscosity syndrome leading to CCF and stroke
- Iron overload
- Only sickle negative blood, which can be identified by negative sickle solubility test should be used
- It should be leucodepleted
- It should be matched for minor E.C &Kell antigens
- If patients baseline Hb is high & dilution of HbS is needed then exchange transfusion (erythrocytopharesis) is needed to prevent hyperviscosity
- Final Hb should not exceed 12g/dL after simple or exchange transfusion. (Goal is 10gm/dL)
- Erythropoietin/ darbepoetin may be used if reticulocyte count is low. Anemia in SCD, can be because of renal dysfunction/ inflammation related BM suppression.
- Fever
- Send complete hemogram, LFT, RFT, blood culture, urine culture, chest X-ray, USG abdomen & SOS CSF analysis
- IV antibiotics for 1week
- For pneumonia a macrolide should be added to cover mycoplasma/ Chlamydia
- Transfuse PRBC if Hb<8gm/dL
- IV fluids- 75-100ml/hr
- SOS Paracetamol
- O2 by face mask
- Pain Management
- Often it is under-rated and under-treated with inexperienced care givers
- Adequate hydration-IV fluids
- Refer “supportive care” section for pain management
- Other options which can be used- Magnesium sulphate / Dapsone/ chlorpromazine/ Morphine/ Ketorulac/ Ibuprofen
- Treatment of precipitating factor
- Oxygen inhalation
- To prevent chronic renal failure
- ACE inhibitors
- Control blood pressure
- Avoid NSAIDS
- Aggressive treatment of UTI & Anemia
- Treatment of priapism
- Explain to boys & ask them to seek early help
- Drink extra fluids, analgesics, warm baths/ soaks
- Attempt to urinate
- Normally episodes are brief& rarely prolonged for > 3 Hrs
- If it does not subside patient must seek immediate medical attention and they must be started on following treatment
- Intravenous hydration
- Analgesia
- Oxygen
- Tab. Pseudoephedrine- <12yers- 1mg/Kg- QID, >12 years- 30-60mg- QID
- Foley's catheterization
- Partial exchange transfusion
- Urology consultation for aspiration of cavernosa/ creation of fistula between glans& corpora cavernosa (Winter procedure)- But this results in permanent impotence
- Further episodes can be prevented by
- Hydroxyurea
- Etilefrine- Alpha adrenergic agonist
- GnRH analogues- Once a month
- Stilbestrol
- Pseudoephedrine
- Bicalutamide- Antiandrogen
- Treatment for acute chest syndrome
- Admit in ICU
- Supplemental oxygen to maintain SpO2 >92%
- SOS- Positive pressure ventilation
- Mechanical ventilation – For rapidly progressive cases
- Adequate pain control
- Incentive spirometry when patient can tolerate
- Strict I/O monitoring and daily weight check- Frusemide if there is fluid overload. IV fluids must be given if patient is unable to maintain adequate oral intake.
- Avoid overhydration
- IV cephalosporinesalong withClarithromycin- 500mg- IV/PO- BD +/- Vancomycin- 1gm- IV- BD
- Antivirals if there is suspicion of H1N1 infection
- Bronchodilators if there is evidence of bronchospasm
- Urgent transfusion
- Partial exchange transfusion (if Hb is >10gm/dL)
- Nitric oxide & steroids – Beneficial in life threatening cases
- Consider thromboprophylaxis
- Treatment for chronic pulmonary hypertension
- Hydroxyurea
- Regular transfusions
- Vasodilators
- Anticoagulation
- Oxygen inhalation
- Sildenafil
- Treatment for papillary necrosis
- Bed rest
- Hydration to maintain high urine output
- Blood transfusion
- If bleeding persists- Vasopressin/ Embolization/ Nephrectomy
- If there is ureteral obstruction- Retrograde ureteral stent
- Treatment of symptomatic avascular necrosis:
- Core decompression
- Osteotomy
- Bone grafting
- Surface orthoplasty
- Joint replacement
- Treatment of ocular complications
- Proliferative retinopathy- Laser photo coagulation and vitrectomy
- Central retinal artery occlusion- Hyper oxygenation and reduction in intraocular pressure
- Treatment of cholelithiasis
- Cholecystectomy is advised even in asymptomatic patients, to avoid subsequent difficulty in distinguishing gall bladder pain from acute painful crisis
- Treatment of acute hepatic crisis
- Supportive care only
- It usually resolves in 3-14 days
- Treatment of lower leg ulcers
- Gentle debridement
- Elastic dressings and leg elevation
- Zinc sulphate
- Topical/ systemic antibiotics
- Skin grafting
- Treatment of splenic sequestration crisis
- Transfusions to restore blood volume with target Hb 8gm/dL
- O2 supplementation
- IV antibiotics
- Splenectomy to prevent recurrence after event has been treated
- Chronic exchange transfusions till age of 2 years
- Treatment of stroke:
- Urgent CT scan to know whether there is infarction/ bleed
- If hemorrhage
- Treatment based on site and amount along with neurosurgical consultation
- If infarct:
- Exchange transfusion with target HbS- 30% and Hb around 10gm/dL
- Treatment as any other stroke- rTPA and aspirin
- Treatment of aplastic crisis:
- IVIg to treat Parvovirus infection
- Transfusions to correct anemia
- Birth control in women
- Modern non estrogen containing intrauterine devices
- Progesterone only OCPs
- Treatment of infertility in men
- Testosterone supplements
- Penile implants for erectile dysfunction
- Psychological issues
- Avoid depression
- Improve family relationships
- Vocational and educational support
- Bone marrow transplantation
- Allogeneic stem cell transplantation from HLA matched sibling
- Cures 85% of children with sickle cell disease less than 16 years of age
- 5-10% die of complications of BMT
- Indications
- Stroke/ Increased TCD velocity
- Impaired neuropsychological function
- Recurrent acute chest syndrome
- Recurrent vaso-occlusive episodes in children less than 16 years who have HLA matched sibling
- Conditioning with Busulfan, cyclophosphamide and ATG
- GVHD prophylaxis with CSA and Methotrexate
- Adding Rapamycin in non-myeloablative conditioning protocols has enabled stable mixed chimerism with full donor erythroid engraftment.
- Unrelated and haplo identical transplants have to be done only on experimental basis in the context of clinical trial.
Newer modalities of treatment
- Senicapoc
- Agent blocking Gardos channel and potassium chloride co transport
- Dose- 10mg/day
- Excellent safety profile. Mild side effects such as nausea and diarrhea
- Increases the hydration of RBCs and hence the deformability
- Agents correcting NO deficiency which is thought to induce vaso-occlusion
- Prostacyclin E5 inhibitors
- Agents inducing HbF synthesis such as butyrate compounds and 5 azacytidine/ Decitabine
- Agents blocking red cell dehydration such as clotrimazole
- Coagulation inhibitors- Tissue factor inhibitors, antiplatelet therapy, anticoagulation
- Agents inhibiting inflammation- Thalidomide, statins etc
- Herbal extracts- Niprisan
Gene therapy
- It is insertion of normal gene into repopulating hematopoietic cells
Special Situations:
- Pregnancy & sickle cell disease
- It is better to avoid pregnancy in sickle cell anemia patients, relative risk of maternal mortality is 6 times higher.
- Steep fall in hemoglobin level is seen
- Exacerbation of folate deficiency
- Painful episodes become more common in last trimester
- Incidence of preeclampsia, UTI, pneumonia, endometritis, VTE, placental abruption and post partum infections is higher
- Fetal complications include abortion, still birth, low birth weight and neonatal death
- If patient wants to conceive/ continue pregnancy
- Give Folic acid- 5mg/kg
- Supplemental iron ifferritin is low
- Discontinue hydroxyureaand iron chelation 3 months prior to pregnancy
- Maintain hemoglobin >7gm/dL
- Avoid inducing labor as it can precipitate sickle cell crisis
- During delivery give supplemental oxygen, maintain hydration and give adequate anesthesia.
- Consider exchange transfusion if there is risk of VTE
- Post delivery thromboprophylaxis must be given.
- If there is previous history of VTE, thromboprophylaxis must be given even during pregnancy.
- Pregnancy in sickle cell trait is uneventful. They have slightly higher risk of UTI.
- Surgery and anesthesia:
- Stress can induce painful crisis
- Transfusions must be given prior to surgery with target hemoglobin of 10gm/dL
- Start IV fluids on the previous day of surgery
- Avoid hypothermia and dehydration during surgery. Also avoid volume overload.
- Post operative pain management must be intensive
- Newborn screening
- Should be done in high risk neonates, as penicillin prophylaxis prevents death due to early sepsis
- Blood samples obtained by heel prick are spotted on to filter paper & tested by electrophoresis or chromatography.
- Prenatal diagnosis
- Done through direct detection of mutation in fetal cells
- Not advised as severity of sickle cell disease cannot be predicted.
- Note: Because parasitized cells sickle more readily leading to sequestration in spleen, sickle cell patients have slight protection against falciparum malaria. Hence this disease is more frequent in countries where malaria is/was endemic.
Related Disorders:
- Sickle cell trait (HbAS)
- Asymptomatic with normal growth & life expectancy
- Prevalence- 8-10%
- No sickle cells are seen in peripheral smear
- Electrophoresis – HbA: HbS = 60:40
- Genetic counseling should be provided to them
- No treatment is needed
- They have higher incidence of pregnancy complications (fetal loss, preeclampsia, prematuredelivery), DVT, papillary necrosis, UTI and renal medullary carcinoma.
- HbSC disease
- Vaso occlusive episodes are present but less severe than SCD
- High incidence of proliferative retinopathy
- Splenomegaly is often present
- Hb – 10-12g/dL
- Peripheral smear- Microcytic anemia, Frequent target cells and folded cells ("pita bread" cells) with intraerythrocytic crystals. Rare sickle cells.
- Electrophoresis – HbS : HbC – 50:50
- Sickle cell - β Thalassemia
- Majority have β + phenotype
- HbA – 3% to 25% (Clinical severity depends on it )
- Peripheral smear- Microcytic hypochromic anemia, target cells and rarely sickle cells
- Presentation is similar to sickle cell anemia, but some can have features of beta thalassemia with splenomegaly.
- Sickle cell with α- Thalassemia
- Less severe anemia
- Peripheral smear - Microcytic hypochromic picture
- Sickle cell- HPFH
- Increased HbF synthesis due to deletional mutation that maintains γ globin gene expression after birth
- They have- HbF – 20-30% and HbA2 - <2.5%
- Hemoglobin concentration – Normal
- Peripheral smear - Macrocytosis & target cells
- Asymptomatic
- Sickle cell disease with Hblepore
- Sickle cell disease with HbD
- Sickle cell disease with HbO Arab
- Sickle cell disease with HbE disease
Figures:
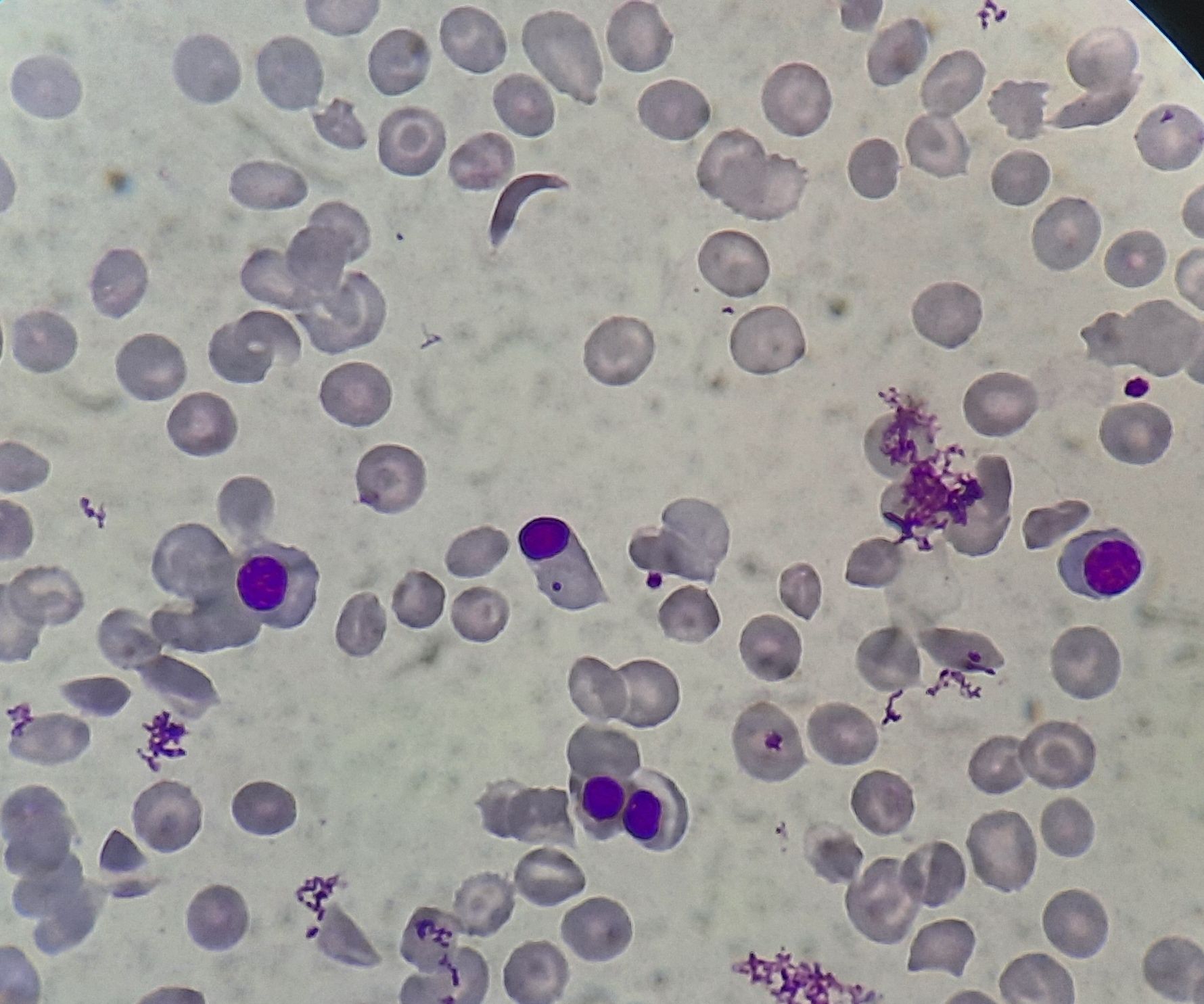
Figure 8.15.1- Sickle cell anemia- Peripheral smear
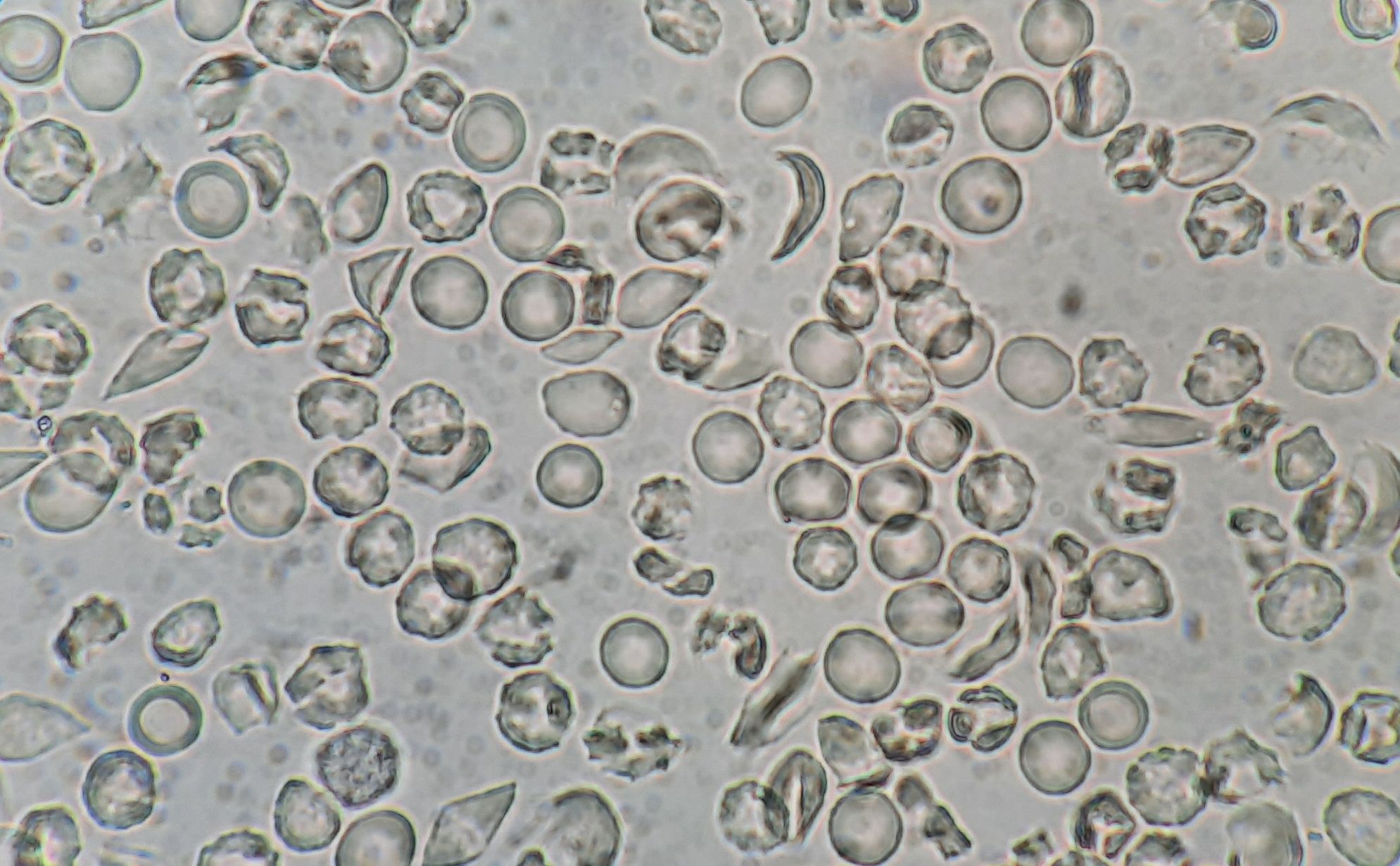
Figure 8.15.2- Sickling test
Recent advances:
Canakinumab in treatment of Sickle cell anemia
Intravascular release of lysed cellular contents from damaged red blood cells activates inflammation in sickle cell anemia patients. This is mainly through release of cytokines such as IL-1beta. Canakinumab causes of blockade of IL-1beta. Hence this was studied in a randomized double blind study. Both group patients continued to receive Hydroxyurea therapy during the trial. Compared with patients in the placebo arm, patients treated with canakinumab had reductions in markers of inflammation and also several clinical parameters.
doi.org/10.1182/blood.2021013674
Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease
Gene therapy using LentiGlobin (lovotibeglogeneautotemcel) was analysed in a recent study which involved 35 patients. All 35 patients had engraftment with rise in median haemoglobin to 11.5gm/dL. Mean HbA levels were at least 40%. All the patients had resolution of severe veno-occlusive crisis. No serious adverse events were seen in any of the patients.
https://doi.org/10.1056/NEJMoa2117175
Alpha haemoglobin-stabilising protein concentration in the red blood cells of patients with sickle cell anaemia with and without hydroxyurea treatment
Alpha haemoglobin-stabilising protein (AHSP) is a key chaperone synthesised in red blood cell (RBC) precursors. Its level was significantly higher in patients treated with hydroxyurea. Researchers observed that AHSP can be used as biomarker for monitoring response to hydroxyurea.
https://doi.org/10.1111/bjh.17728
Pyruvate kinase activator mitapivat in sickle cell disease
Mitapivat increases adenosine triphosphate levels and decreases the 2,3-diphosphoglycerate concentration. Both changes have therapeutic potential for patients with SCD. Mitapivat was well tolerated at all dose levels, with the most common treatment-emergent adverse events being insomnia, headache, and hypertension. Mean hemoglobin increase at the 50 mg BID dose level was 1.2 g/dL.
https://doi.org/10.1182/blood.2022015403
Rivipansel for of sickle cell vaso-occlusive crisis
Rivipansel is a predominantly E-selectin antagonist. Present study examined use of this agent in sickle cell vaso-occlusive crisis. In the full analysis population, the median time to readiness for discharge, was not different between rivipansel and placebo, nor were differences seen in secondary end points of time to discharge, time to discontinuation of IV opioids, and cumulative IV opioid use. Mean soluble E-selectin decreased 61% from baseline after the loading dose in the rivipansel group, while remaining unchanged in the placebo group.
https://doi.org/10.1182/blood.2022015797
Hydroxyurea for secondary stroke prevention in children with sickle cell anemia
Present study tested the hypothesis that fixed oral moderate-dose hydroxyurea (20 mg/kg per day) for initial treatment of secondary stroke prevention results in an 80% relative risk reduction of stroke or death when compared with fixed oral low-dose hydroxyurea (10 mg/kg per day). The trial was stopped early owing to no clinical difference in the incidence rates of the primary outcome measure. The incidence rates of recurrent strokes were 7.1 and 6.0 per 100 person-years in the low- and moderate-dose groups, respectively.
https://doi.org/10.1182/blood.2022016620
Factor XII contributes to vaso-occlusion in sickle cell disease
Present study investigated whether FXII contributes to the prothrombotic and inflammatory complications associated with SCD. Study found that when compared with healthy controls, patients with SCD exhibit increased circulating biomarkers of FXII activation that are associated with increased activation of the contact pathway. We also found that FXII, but not tissue factor, contributes to enhanced thrombin generation and systemic inflammation observed in sickle cell mice challenged with tumor necrosis factor α.
https://doi.org/10.1182/blood.2022017074
CRISPR-Cas9 Editing of the HBG1 and HBG2 Promoters to Treat Sickle Cell Disease
OTQ923, a CRISPR-Cas9-edited product of hematopoietic stem cells, was developed to increase fetal hemoglobin expression. Preclinical experiments showed successful editing of stem cells, leading to sustained high levels of fetal hemoglobin with no off-target mutations. In a clinical study, three severe sickle cell disease patients received OTQ923, resulting in stable induction of fetal hemoglobin (19.0 to 26.8% of total hemoglobin) and clinical improvement, suggesting CRISPR-Cas9 disruption as an effective strategy.
https://doi.org/10.1056/NEJMoa2215643
Increased retention of functional mitochondria in mature sickle red blood cells is associated with increased sickling tendency
The study identified abnormal retention of mitochondria in mature red blood cells (RBC) in sickle cell anemia (SCA). SCA patients were classified based on the percentage of mature RBC with mitochondria. Functional mitochondria were detected in sickle mature RBC but not in healthy RBC. Increased mitochondrial reactive oxygen species were observed in sickle RBC retaining mitochondria, leading to higher levels of reactive oxygen species, greater Ca2+ levels, lower CD47, and increased phosphatidylserine exposure. The SCA group with a high percentage of mitochondria retention in mature RBC exhibited lower hematocrit, reduced RBC deformability, and increased propensity for sickling under deoxygenation, suggesting a role in accelerating RBC senescence and hemolysis.
https://doi.org/10.3324/haematol.2023.282684
Neurofilament light chain: A potential biomarker for cerebrovascular disease in children with sickle cell anaemia
In a study involving Ugandan children with sickle cell anaemia (SCA), researchers explored the relationship between plasma-based brain biomarkers and cerebral infarcts detected by magnetic resonance imaging (MRI) and transcranial Doppler (TCD) arterial velocity. The study included a 4-plex panel of brain injury biomarkers, with a focus on neurofilament light chain (NfL). Children with SCA and MRI-detected infarcts had higher NfL levels compared to those without infarcts, and elevated NfL was associated with abnormal TCD velocity. The findings suggest that plasma-based NfL levels may have potential utility in identifying SCA-related cerebrovascular injury. Prospective studies are needed for confirmation.
https://doi.org/10.1111/bjh.19036
Oral famotidine reduces the plasma level of soluble P-selectin in children with sickle cell disease
In a prospective, non-comparative study, oral famotidine, a histamine type 2 receptor antagonist, was administered to children with sickle cell disease (SCD). After 29 days of treatment, famotidine significantly reduced plasma P-selectin levels in the patients (53.2 ng/mL vs. 69.9 ng/mL). No adverse events related to famotidine were observed, and the study suggests that further randomized controlled trials are needed to assess the efficacy of famotidine in preventing vaso-occlusion in SCD.
https://doi.org/10.1111/bjh.19111
Hydroxyurea is associated with later onset of acute splenic sequestration crisis in sickle cell disease
This study examined the association between hydroxyurea (HU) treatment and the onset of acute splenic sequestration crisis (ASSC) in patients with sickle cell disease (SCD). Data from the ESCORT-HU study, including 7309 patient-years of observation, revealed that the median age at first ASSC episode was significantly later in patients receiving HU (8.0 years) compared to those not receiving HU (median age of 4.8 years at HU initiation). These findings suggest a potential delay in the onset of ASSC with HU treatment, possibly due to improved spleen perfusion and function.
https://doi.org/10.1002/ajh.27214
Riociguat in patients with sickle cell disease and hypertension or proteinuria
Riociguat is a stimulator of soluble guanylate cyclase (sGC), an enzyme in the cardiopulmonary system and the receptor for nitric oxide (NO). It is used to treat adults with chronic thromboembolic pulmonary hypertension. In a phase 1–2, randomised, double-blind, placebo-controlled trial, riociguat was evaluated for its safety and efficacy in patients with sickle cell disease. Riociguat significantly reduced systemic blood pressure compared to placebo. The findings supported the safety of riociguat in patients with sickle cell disease.
https://doi.org/10.1016/S2352-3026(24)00045-0
Exagamglogene Autotemcel for Severe Sickle Cell Disease
In a phase 3 study, exagamglogene autotemcel (exa-cel) demonstrated efficacy in reducing vaso-occlusive crises in patients aged 12 to 35 with sickle cell disease. Patients underwent CRISPR-Cas9 gene editing of autologous CD34+ hematopoietic stem and progenitor cells (HSPCs) targeting the BCL11A erythroid-specific enhancer region. Following myeloablative conditioning with busulfan, patients experienced engraftment of neutrophils and platelets. The primary endpoint of freedom from severe vaso-occlusive crises for at least 12 consecutive months was achieved by 97% of patients, with all patients free from hospitalizations for vaso-occlusive crises for the same duration. The safety profile was consistent with myeloablative conditioning and autologous HSPC transplantation, with no reported cases of cancer.
https://doi.org/10.1056/NEJMoa2309676
Sputum interleukin-6 level as a marker of severity during acute chest syndrome in children with sickle cell disease
In a prospective observational study involving 26 children with sickle cell disease (SCD) experiencing 30 episodes of acute chest syndrome (ACS), sputum interleukin-6 (IL-6) levels measured within the first 72 hours of hospitalization were evaluated as markers of ACS severity. The study found that higher sputum IL-6 levels correlated significantly with ACS severity indicators such as oxygen requirement of ≥2 L/min, prolonged ventilation (≥5 days), bilateral or extensive chest X-ray opacities, and the need for erythrocytapheresis. These findings suggest that sputum IL-6 could potentially serve as a useful biomarker for identifying ACS patients who may benefit from targeted anti-inflammatory treatments like tocilizumab.
https://doi.org/10.1111/bjh.19561
The influence of voxelotor on cerebral blood flow and oxygen extraction in pediatric sickle cell disease
A pilot study evaluated the cerebral hemodynamic effects of voxelotor, a sickle hemoglobin polymerization inhibitor, in children with sickle cell anemia (SCA). Using noninvasive optical techniques, the study measured cerebral blood flow (CBFi), oxygen extraction fraction (OEF), and blood hemoglobin (Hb) levels. Results showed significant decreases in both OEF and CBFi by 4 weeks of treatment, which persisted through 12 weeks. These changes were correlated with increases in Hb, suggesting that voxelotor may alleviate cerebral hemodynamic impairments in pediatric SCA patients.
https://doi.org/10.1182/blood.2023022011
Oral carbon monoxide–releasing molecule protects against acute hyperhemolysis in sickle cell disease
This study explored the protective effects of the CO-releasing molecule CORM-401 against acute hyperhemolysis-induced organ damage in sickle cell disease (SCD). CORM-401 was shown to prevent endothelial activation and inflammation in vitro by modulating NF-κB and Nrf2 pathways. In SCD mice, it effectively reduced organ damage in the lung, liver, and kidney. These findings suggest CORM-401 as a potential preventive treatment for high-risk SCD patients.
https://doi.org/10.1182/blood.2023023165
Hydroxyurea dose optimisation for children with sickle cell anaemia
The REACH trial examined hydroxyurea treatment in children with sickle cell anaemia in sub-Saharan Africa over 8 years. 606 children received hydroxyurea, starting with a fixed dose of 17.5 (±2.5) mg/kg per day for 6 months, followed by dose escalation up to 20-35 mg/kg per day based on tolerance. The current mean dose is 28.2 (SD 5.2) mg/kg per day. This treatment showed significant improvements in haemoglobin levels, fetal haemoglobin, and reduced rates of vaso-occlusive episodes, acute chest syndrome, infections, and serious adverse events. Dose escalation to the maximum tolerated dose (MTD) with optimization significantly enhanced clinical responses and outcomes without increasing toxicities, supporting hydroxyurea's long-term efficacy and safety in this population.
https://doi.org/10.1016/S2352-3026(24)00078-4
Safety and efficacy of L-Glutamine in reducing the frequency of acute complications among patients with sickle cell disease
This randomized controlled trial assessed the safety and efficacy of L-glutamine in reducing vaso-occlusive crises (VOCs) and improving cerebral arterial blood flow in 60 children with sickle cell disease (SCD). Over 24 weeks, patients receiving glutamine had significantly fewer VOCs and hospitalizations compared to those on standard care. There was also a trend toward decreased VOC severity, and glutamine appeared to normalize cerebral arterial blood flow velocities. The study suggests that L-glutamine may reduce VOC frequency and severity, with potential benefits for cerebral blood flow in SCD children.
https://doi.org/10.1007/s00277-024-05877-8
Azathioprine/hydroxyurea preconditioning prior to nonmyeloablative matched sibling donor hematopoietic stem cell transplantation in adults with sickle cell disease
In this prospective study, 20 adult sickle cell disease (SCD) patients underwent nonmyeloablative matched sibling donor hematopoietic stem cell transplantation with azathioprine/hydroxyurea preconditioning. The regimen led to high one-year overall and event-free survival rates of 95%. The mean donor myeloid and T-cell chimerism levels were 95.2% and 67.3%, respectively. Only one patient experienced graft failure and subsequently died, while the rest achieved corrected SCD phenotypes, allowing discontinuation of sirolimus. Preconditioning with azathioprine/hydroxyurea improved outcomes with minimal toxicity, demonstrating its potential to reduce graft rejection and improve donor chimerism.
https://doi.org/10.1002/ajh.27360
An Initiative of
Veenadhare Edutech Private Limited
1299, 2nd Floor, Shanta Nivas,
Beside Hotel Swan Inn, Off J.M.Road, Shivajinagar
Pune - 411005
Maharashtra – India

howitreat.in
CIN: U85190PN2022PTC210569
Email: admin@howitreat.in
Disclaimer: Information provided on this website is only for medical education purposes and not intended as medical advice. Although authors have made every effort to provide up-to-date information, the recommendations should not be considered standard of care. Responsibility for patient care resides with the doctors on the basis of their professional license, experience, and knowledge of the individual patient. For full prescribing information, including indications, contraindications, warnings, precautions, and adverse effects, please refer to the approved product label. Neither the authors nor publisher shall be liable or responsible for any loss or adverse effects allegedly arising from any information or suggestion on this website. This website is written for use of healthcare professionals only; hence person other than healthcare workers is advised to refrain from reading the content of this website.