
howitreat.in
A user-friendly, frequently updated reference guide that aligns with international guidelines and protocols.
Suspected Bleeding Disorder
Indications for hemostatic evaluation:
- Suspected bleeding tendency
- Bleeding tendency discovered in any family member
- Abnormal coagulation assays report during routine/pre surgery evaluation
- Unexplained diffuse bleeding during or after surgery or following trauma
Differential Diagnosis of bleeding problems
- No bleeding disorder- Symptoms do not reflect bleeding disorder and have another explanation. (Ex: Surgical bleed which is not due to a bleeding disorder)
- Possible bleeding disorder- The laboratory findings are non-diagnostic and bleeding history is considered equivocal Ex: Unexplained serious bleed with one surgical procedure, unexplained menorrhagia without other bleeding problems.
- Definite bleeding disorder undefined or indeterminate type- Bleeding history is consistent with a bleeding disorder however lab findings are non diagnostic or not available.
- Definite bleeding disorder with a defined cause – Symptoms and lab findings are considered diagnostic of a bleeding disorder
Classification of diseases of hemostasis
- Inherited
- Deficiency of coagulation factor- 8, 9, 2, 5, 7, 9, 11, 13, VWF.
- Platelet function disorders- Glanzmanthrombasthenia, Bernard Soulier syndrome, platelet granule disorders.
- Fibrinolytic disorders- Antiplasmin deficiency, plasminogen activator inhibitor 1 deficiency.
- Vascular- Hemorrhagic telangiectasia.
- Connective tissue disorders – EhlerDanlos syndrome.
- Acquired
- Liver diseases- Cirrhosis, acute hepatic failure, liver transplantation, TPO deficiency.
- Renal failure
- Vitamin K deficiency- Malabsorption syndrome, hemorrhagic disease of new born, prolonged antibiotic therapy, malnutrition
- Hemorrhagic disorders- AML particularly APML, myelodysplasia, monoclonal gammopathy, essential thrombocythemia
- Acquired antibodies against coagulation factor- Neutralizing antibodies against F V, VIII, XII which leads to accelerated clearance of factor Ex: Acquired VWD, Hypoprothrombinemia associated with APLA.
- DIC- Sepsis, malignancies, trauma, obstetric complications
- Drugs- Anti platelet agents, anticoagulants, antithrombins, thrombolytic, hepatotoxic agents, Ginkgo biloba
- Vascular- Non palpable purpura (Senile, solar, factitious, use of steroids, vitamin C deficiency, child abuse, amyloidosis etc)
- Massive blood transfusion
- Hypothyroidism
- Other conditions causing thrombocytopenia
Investigations
- Complete hemogram including peripheral smear
- Screening tests- PT, APTT, TT, Fibrinogen
- Coagulation proteins need to decrease to different low levels before the various screening tests show an abnormality.Ex: Most commercial APTT detect decrease in factor VIII when protein level decreases to 35%-45% of normal and XII and HMWK at levels of 10-15% of normal.PT is prolonged once F VII levels are below 35%
- Mixing studies-
- If PT/APTT are prolonged.
- Test correction in mixing study indicates a deficiency state
- Lack of correction indicates specific factor inhibitor or lupus anticoagulant.
- Factor VIII inhibitors are time and temperature dependent. Hence test must be repeated after incubating the patient plasma with normal standard plasma at 37 degree C for 1-2 hrs)
- Factor assays- Based on PT, APTT and mixing study results.
- Platelet function test
- RFT and LFT
- D-Dimer- If DIC is suspected
- Thyroid function test-hypothyroidism can cause increased bleeding
- Bleeding time and clotting time- They are not recommended now
If asymptomatic, investigate the patient if
- PT >5 Sec than control
- APTT >8 Sec than control
- Immediate first step is to do mixing study to differentiate factor deficiency from presence of inhibitors
Approach to diagnosis:
PT | APTT | TT | Fibrinogen | Platelet count | Differential diagnosis |
Normal | Normal | Normal | Normal | Normal | Platelet dysfunction- Congenital/ acquired PAI-1 deficiency Alpha 2 AP deficiency |
Prolonged | Normal | Normal | Normal | Normal | Factor 7 deficiency |
Normal | Prolonged | Normal | Normal | Normal | Factor 8, 9, 11, 12, prekallirein or HMWK deficiency |
Prolonged | Prolonged | Normal | Normal | Normal | Vitamin K deficiency |
Prolonged | Prolonged | Prolonged | Normal | Normal | Heparin (Large amounts) |
Normal | Normal | Normal | Normal | Low | Thrombocytopenia |
Prolonged | Prolonged | Normal | Normal/Abnormal | Low | Massive transfusion |
Prolonged | Prolonged | Prolonged | Low | Low | DIC |
Note: Deficiency of factor 12, HMWK, and Prekallikrein do not have with bleeding tendency
Thrombocytopenia
Introduction:
- It is a condition in which platelet count is less than 1.5 lac/cmm.
- But abrupt drop in platelet count, with platelet count still above 1.5 lac /cm, is significant and should be investigated
- Platelet count of 1 lac-1.5 lac/cmm with patient being stable for >6 months, does not indicate disease and does not need further evaluation.
- Following are thrombocytopenic emergencies. These conditions have to be excluded first, before considering other causes of thrombocytopenia, as they can be fatal if not treated urgently.
- Heparin induced thrombocytopenia
- Thrombotic thrombocytopenic purpura
- Disseminated intravascular coagulation
- Catastrophic antiphospholipid antibody syndrome
- Primary immune thrombocytopenia with bleeding
- Post-transfusion purpura
Causes of thrombocytopenia
- Pseudo thrombocytopenia.
- Platelet agglutination.
- Platelet satellitism.
- Antiphospholipid antibodies.
- GpIIa, IIIa antibodies.
- Giant platelets.
- Impaired platelet production
- Congenital (Auto dominant)
- MYH9 related- May Hegglin anomaly, Fechtner syndrome, Epstein syndrome, Sebastin syndrome.
- Mediterranean macro thrombocytopenia
- Familial platelet syndrome with predisportion to AML
- Thrombocytopenia with linkage to chromosome 10
- Paris Trousseau syndrome
- Thrombocytopenia with radial synostosis
- Congenital (Autosomal Recessive)
- Congenital amegakaryocytic thrombocytopenia
- Thrombocytopenia with absent radii syndrome
- Bernard Soulier syndrome
- Gray PL syndrome
- X- Linked thrombocytopenia
- Wiskott Aldrich syndrome
- X-linked thrombocytopenia with dyserythrocytosis
- Acquired
- Marrow infiltration- leukemia, lymphoma, myelofibrosis
- Infectious disease- HIV, Parvo virus, Dengue, viral fever, malaria, CMV, E. Coli, shigella, mycobacteria, Rickettsia
- Radiotherapy, Chemotherapy.
- Folic acid, Vitamin- B-12 deficiency
- Drug induced- Sulfa antibiotics, valproic acid
- Paroxysmal nocturnal hemoglobinuria
- Aplastic anemia
- MDS
- HLH
- Acquired pure amegakaryocytic thrombocytopenia (May be associated with other autoimmune disorders, rarely may progress to MDS/ aplastic anemia, Treated with immunosuppressive therapy -ATG with Cyclosporine)
- Alcoholism
- Congenital (Auto dominant)
- Accelerated platelet destruction
- Immune mediated
- Immune thrombocytopenic purpura
- Secondary immune thrombocytopenia- Infection (Hepatitis C, H. Pylori), pregnancy related, lymphoproliferative disorders, collagen vascular disease. Ex: SLE, RA, APLA, vaccination related
- Allo immune thrombocytopenia.
- Neonatal thrombocytopenia purpura
- Post transfusion purpura
- Drug induced thrombocytopenia
- Non immune mediated
- Thrombotic microangiopathy – TTP, HUS, DIC, Eclampsia/HELLP syndrome, catastrophic APLA
- Septicemia
- Infections- Dengue, HIV, Malaria
- Kasabach Meritt syndrome (Large Hemangioma)
- Platelet destruction by artificial surfaces
- Gestational thrombocytopenia
- Splenomegaly/ hypersplenism
- Massive transfusion (Hemodilution related)
- Post-surgical thrombocytopenia
- Heparin induced thrombocytopenia
- Other drug induced thrombocytopenia
- Extracorporeal circuit/ Intravascular devices-
- Cardiopulmonary bypass surgery (Often platelet count falls to around 50,000/cmm, associated with platelet dysfunction, No benefit of transfusion of platelets)
- Intra-aortic balloon pumps
- Extracorporeal membrane oxygenation
- Decreased platelet survival associated with cardiovascular disease- congenital cyanotic disease, valular heart disease, cardiomyopathy
- Hypothermia related thrombocytopenia
- Immune mediated
Clinical manifestations of thrombocytopenia (Spontaneous bleeding is seen only when platelet count drops to less than 20,000/cmm)
- Purpura- skin (Dry purpura), Oral mucosal bleeding (Wet purpura). Wet purpura is a sign of impending serious internal bleeding such as intracranial bleeding.
- Increased PV bleeding, hematochezia, malena, epistaxis, hemoptysis, hematemesis
Investigations
- CBC- To know whether patient has isolated thrombocytopenia or pancytopenia.
- Peripheral smear examination:
- Pseudo thrombocytopenia (EDTA induced platelet clumping, giant platelets, platelet satellitism etc)
- RBC fragments suggest microangiopathy- TTP, HUS, or DIC
- Moderately enlarged platelets- Seen in ITP and other conditions with accelerated platelet destruction
- Small platelets- Seen in Wiskott aldrich syndrome
- Macrocytosis and hypersegmented neutrophils are seen in megaloblastic anemia
- Target cells are seen in liver diseases
- Abnormal WBCs are seen in acute leukemias and MPN
- Dysplastic changes such as Pelger Huet anomaly suggest myelodysplastic syndrome
- Leucoerythroblastic reaction is suggestive of bone marrow infiltration or myelofibrosis
- Toxic changes and granulocyte "left shift" suggest septicemia
- Atypical lymphocytes suggest viral infection such as dengue
- Parasites Ex: malaria
- White cell inclusions (Dohle bodies) suggest hereditary macrothrombocytopenia (MYH9 related)
- LFT
- RFT
- LDH
- Blood cultures- Bacteremia, fungemia
- ANA test
- Direct antiglobulin test- To exclude immune hemolysis accompanying ITP (Evans syndrome)
- Coagulation assays- APTT,PT (INR), TT, fibrinogen, D-dimer assay- To rule out DIC
- Lupus Anticoagulant assay (nonspecific APL antibody syndrome inhibitor), anticardiolipin and anti beta2 Glycoprotein I assays- For APLA
- Serum protein electrophoresis, IgG,IgM,IgA levels: Monoclonal in case of ITP associated with lymphoproliferative disorder and polyclonal in case of chronic hepatitis
- HIV serologic study
- BM aspiration, biopsy- Assesses megakaryocyte numbers and morphology; exclude primary BM disorder, Metastasis, Goucher disease, leukemia, megaloblastic anemia
- USG abdomen- to rule out chronic liver disease and to check for organomegaly and enlarged abdominal/pelvic lymph nodes
Specialized tests
- Platelet associated IgG- It is not useful due to high prevalence in normal population as well
- Drug dependent increase in platelet associated IgG- It is specific assay for drug induced ITP
- Drug dependent platelet activation test (e. g. Platelet serotonin release assay) or PF4 heparin (or PF4 –polyanion) ELISA- Done for diagnosis of heparin induced thrombocytopenia
- Radionuclide platelet lifespan study with imaging (Ex: 111In platelet survival study)- Helps in defining the mechanism of thrombocytopenia, also helps to identify an accessory spleen in post-splenectomy patients)
- Reticulated platelet count:
- Counting of newly released circulating platelets which have RNA
- Helps in discriminating between thrombocytopenia due to BM failure (RPC is decreased) and hyperdestructive thrombocytopenia (RPC is increased.
- Normal -3-20%
Diagnostic algorithm (For neonatal thrombocytopenia- Refer consultative hematology section)
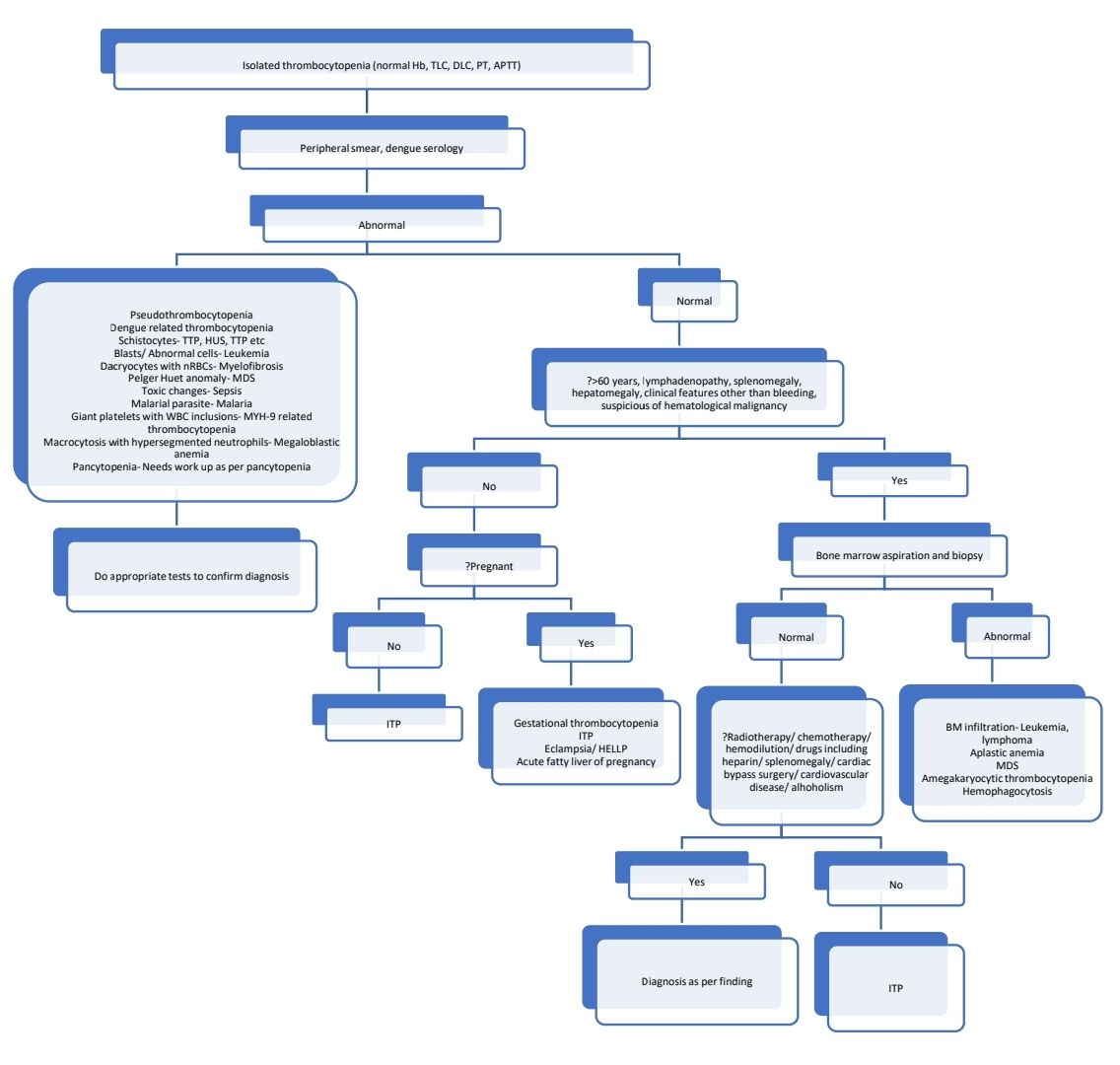
General principles of management
- Avoid drugs that impair hemostasis –Ex: Alcohol, antiplatelet agents, anticoagulants
- Avoid invasive procedure including IM injections
- Stop suspected drug causing thrombocytopenia
- If life threatening bleed – Transfuse platelets irrespective of mechanism of thrombocytopenia
- Do not give prophylactic platelets to patients who are stable (ITP, aplastic anemia, MDS etc)
- Keep a cut-off PL count > 50000/cmm for invasive procedures like – Thoracentesis, paracentesis and liver biopsy.
- No platelets should to be given in patients strongly suspected or confirmed HIT, TTP, or HUS (As thrombotic complications may increase)
Causes of thrombocytopenia with anemia
- TTP
- Evan syndrome
- DIC
- HELLP syndrome
- HUS
- Drugs - Quinine, Simvastatin, interferon, calcineurin inhibitors, clopidogrel
- Malignant hypertension.
- Infections
- Viral-CMV, Adeno, HSV, AIDS, HCV
- Bacterial: Meningococcus, Pneumococcus, H. Pylori
- Fungal
- Autoimmune: Lupus nephritis, acute scleroderma, Autoimmune hepatitis
- Malignancy
- Catastrophic APLA
- Progressive systemic sclerosis
- Heparin induced thrombocytopenia
- Lymphoma – HD, CLL
- Liver disease
- MDS, leukemia, Myelofibrosis, aplastic anemia, megaloblastic anemia
Causes of thrombocytopenia with thrombosis
- Heparin induced thrombocytopenia
- Malignancies
- DIC
- TTP
- HUS
- SLE
- Warfarin induced gangrene
- Sepsis
- APLA
- PNH
Figures:
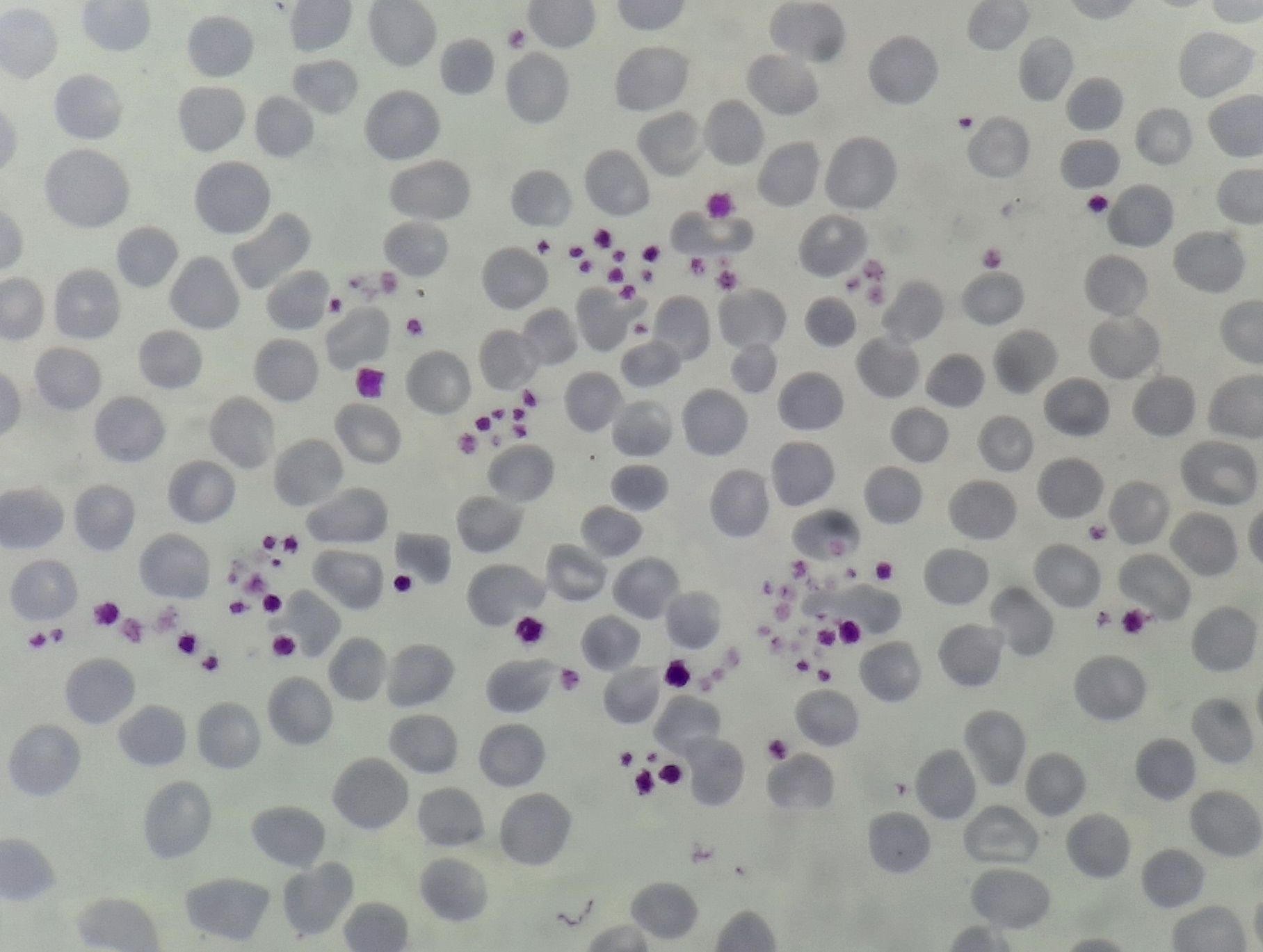
Figure 12.10.1- Pseudothrombocytopenia due to platelet clumps
An Initiative of
Veenadhare Edutech Private Limited
1299, 2nd Floor, Shanta Nivas,
Beside Hotel Swan Inn, Off J.M.Road, Shivajinagar
Pune - 411005
Maharashtra – India

howitreat.in
CIN: U85190PN2022PTC210569
Email: admin@howitreat.in
Disclaimer: Information provided on this website is only for medical education purposes and not intended as medical advice. Although authors have made every effort to provide up-to-date information, the recommendations should not be considered standard of care. Responsibility for patient care resides with the doctors on the basis of their professional license, experience, and knowledge of the individual patient. For full prescribing information, including indications, contraindications, warnings, precautions, and adverse effects, please refer to the approved product label. Neither the authors nor publisher shall be liable or responsible for any loss or adverse effects allegedly arising from any information or suggestion on this website. This website is written for use of healthcare professionals only; hence person other than healthcare workers is advised to refrain from reading the content of this website.