
howitreat.in
A user-friendly, frequently updated reference guide that aligns with international guidelines and protocols.
Von Willebrand Disease
Introduction:
- It is the most common inherited disorder of bleeding in human beings
Von Willebrand factor
- It is a protein synthesized by endothelial cells and megakaryocytes
- It is stored in Webel Palade bodies of endothelial cells and alpha granules of platelets.
- It acts as a carrier protein for factor VIII and also prevents its degradation.
- It facilitates platelet adhesion to vascular subendothelium. At one end it binds to collagen and at other end it binds to platelets.
- Its release from endothelial stores in induced by epinephrine, histamine and vasopressin
- Chronic elevation of VWF occurs as a part of “acute phase response” to injury, inflammation, infection etc.,
- Individuals with blood group ‘O’ have vWF levels 25-30% lower than those with non ‘O’ groups
- Gene encoding vWF is located on chromosome 12
Epidemiology:
- Incidence: 1 in 100 individuals
- Incidence of clinically significant vWD- 1 in 1000.
Common clinical types and their features:
- Type 1: Partial deficiency due to impaired secretion (because of intracellular retention of vWF) or increased clearance.
- Type 2A: vWF variants with loss of high –molecular weight multimers (due to decreased multimerization or increased susceptibility to ADAMTS13) and decreased platelet dependent function
- Type 2B: vWF variants (gain of function mutations) with loss of high molecular weight multimers caused by increased affinity for platelet glycoprotein lb.Causes spontaneous binding of VWF to platelets forming platelet aggregates which are removed from the circulation
- Type 3:Severe deficiency (virtually complete absence) of vWF
- Platelet type: Reduction in large multimers caused by “consumption” by platelets.
- Type 2M- vWF variants with decreased platelet dependent function not associated with the loss of high molecular weight multimers. Very rare disorder with occasional case reports.
- Type 2 N- vWF variants with decreased affinity for factor 8. In this VWF antigen, RIPA, RICOF and vWF multimeric structure are normal but factor 8 levels are disproportionately low. These patients present like haemophilia with frequent joint bleeds.
Features | Type 1 | Type 2A | Type 2B | Type 3 | Platelet –Type |
Inheritance | Autosomal dominant | Autosomal dominant | Autosomal dominant | Autosomal recessive | Autosomal dominant |
Bleeding time | Normal or prolonged | Prolonged | Prolonged | Prolonged | Prolonged |
Factor VIII activity in plasma | Normal or reduced | Normal or reduced | Normal or reduced | Reduced | Normal or reduced |
vWF antigen | Normal or reduced | Normal or reduced | Normal or reduced ; increased affinity for platelet | Reduced | Normal or reduced ; increased affinity for platelet |
vWFmultimeric analysis | Normal | Absence of large and ntermediate sized multimers | Absence of large multimers | Variable abnormalities ;preponderance of small multimers or absent multimers | Reduction in large multimers caused by “consumption” by platelets |
Ristocetin cofactor activity | Normal or reduced | Reduced | Normal or reduced | Reduced | Normal or reduced |
VWF:RCo (IU/dL) | <30 | <30 | <30 | <5 | - |
Ristocetin induced platelet aggregation | Normal or diminished | Diminished | Increased aggregation at low ristocetin concentration | Markedly diminished | Hyperaggregation with patients platelets, normal plasma and low concentration of ristocetin |
vWF in platelets | Normal or reduced | Normal or absence of large and intermediate sized multimers | Normal | Absent | Normal |
Ancillary findings | DDAVP usually produce significant increase in plasma VIIIc and vWF | DDAVP produces rise in factor VIIIc, but functional vWF increase is variable and may be of short duration | Variable response to DDAVP, with intravascular platelet aggregation and thrombocytopenia in some cases ; ristocetin cofactor levels and ristocetin induced platelet aggregation enhanced in presence of patient’s plasma ; cryoprecipitate does not aggregate platelets in vitro unless ristocetin is added | Response to DDAVP lacking ; endothelial vWF absent ; carriers difficult to detect | Transfusion of vWF or DDAVP may produce intravascular platelet aggregation and thrombocytopenia cryoprecipitate produces in vitro platelet aggregation |
Clinical Features:
- Superficial bruising
- Epistaxis
- Menorrhagia – 20% of women with menorrhagia have vWD
- GI haemorrhage- Association with hereditary haemorrhagic telangiectasia has been reported.
- Gingival bleed
- Postpartum haemorrhage
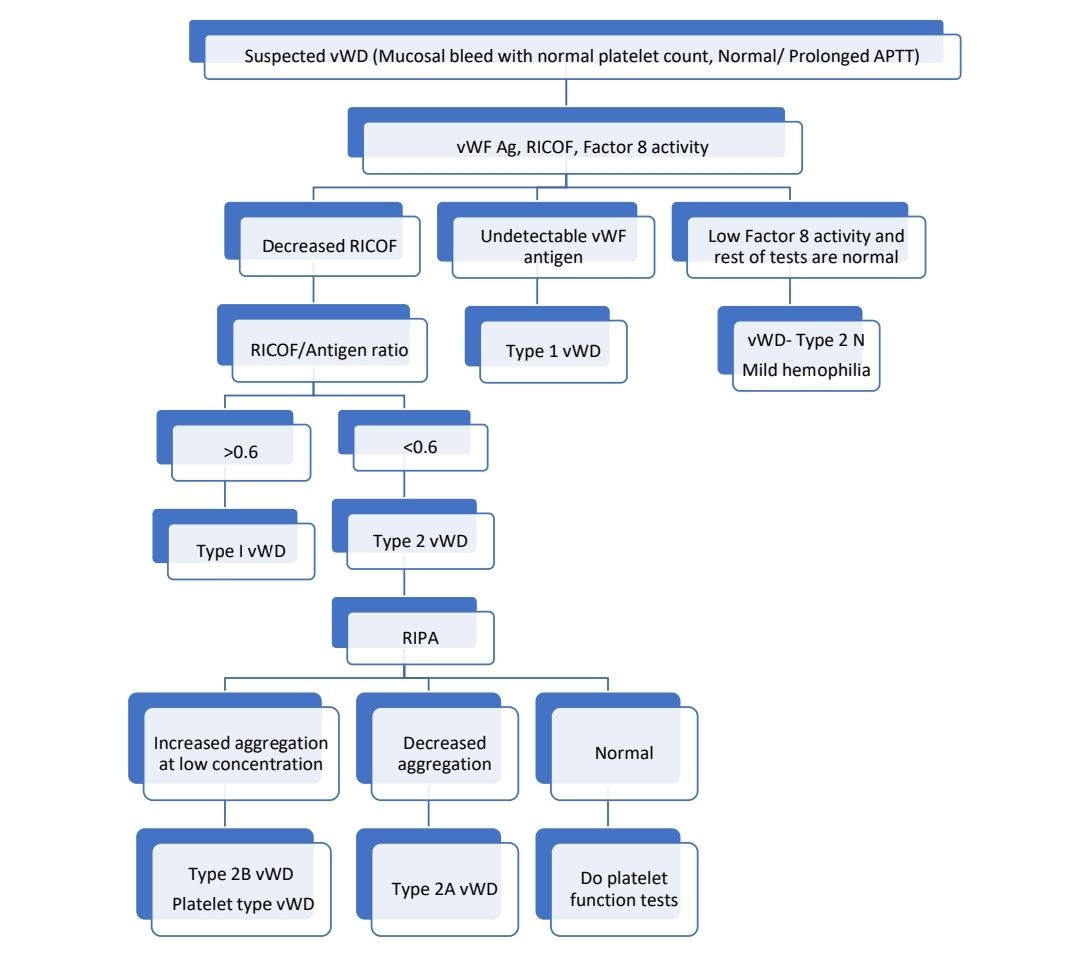
Investigations: No single test is adequate to diagnose vWD by itself.
- Bleeding time – Prolonged
- PT- Normal
- APTT- Normal or Slightly prolonged secondary to decrease in Factor 8 levels.
- Levels of vWF antigen (Done by ELISA)- Reduced
- Factor VIII activity – Decreased
- Ristocetin co-factor assay test –
- It measures vWF activity
- Normal multimeric vWF causes agglutination of platelets in presence of restocetin
- Ability of patient's plasma to aggregate normal platelets (Fresh/ Fixed) in the presence of Ristocetin is compared with that of normal pooled plasma specimen.
- Restocetin brings in conformational change in vWF and facilitates binding of platelet GpIb to vWF.
- >50% is considered as normal (O Blood group may have slightly lower values)
- vWF ristocetin/vWF antigen ratio- <0.6 suffests qualitative defect in vWF i.e., type 2 disease
- Other test to assess vWF activity is collagen binding test
- VWF multimeric assay
- It is done using SDS agarose gel electrophoresis technique to separate vWF into bands
- Loss of large and intermediate multimers is seen in type 2A and 2B.
- Ristocetin induced platelet aggregation analysis (RIPA)
- Aggregation of patient’s platelets (i.e. platelet rich plasma) is tested in the presence of different concentrations of ristocetin.
- In normal individuals, low concentrations of ristocetin are not sufficient to initiate vWF dependent platelet aggregation. Platelet aggregation at low levels of restocetin concentration suggests pathological enhancement of vWF-GP-Ib interaction which is characteristic of type 2B vWD and platelet type pseudo VWD.
- It is used for differentiating type 2A from type 2B.
- This test is generally done after diagnosis of VWD is made using RICOF.
- vWF mutation analysis
- Needed in some cases
- Helps to clarify disease type and risk of disease inheritance
- Facilitates prenatal diagnosis
- Necessary to differentiate
- VWD type 2N and haemophilia A
- vWD type 2B and platelet type VWD
Criteria for Diagnosis: VWF activity less than 30IU/dL
Pretreatment work up
- Assess severity of bleeding phenotype
- Screen for HIV, HBV and HCV
- Baseline iron studies
- Gynaec evaluation for patients with menorrhagia
- Perform desmopressin challenge test for all type 1 and subset of Type 2 patients.
Treatment:
- Desmopressin
- It causes release of stored vWF from endothelial cells.
- Effective only in Type 1 vWD
- Type III do not respond due to lack of gene.
- Strictly contraindicated in in type 2B
- Should not be used in patients with unstable coronary artery disease, as there can be large multimer vWF induced platelet aggregation.
- Relatively inexpensive, easily available and decreased risk of plasma derived products.
- Causes fluid retention- Hence after the dose fluid intake must be restricted for 24 hours.
- Avoid in children aged less than 2 years and elderly aged more than 70 years, due to high risk of hyponatremia.
- Dose for small haemorrhages - 0.3 µg/kg – Intranasal / subcutaneous/ IV infusion over 30min or 300microgm intranasally
- Immediately after diagnoses test dose of desmopressin must be given and increase in level of vWF must be documented. (Measurements are done at 1hr and 4hr after administration)
- Factor VIII concentrates containing VWF
- Used when there is severe bleeding/ surgery is planned
- Humate – P – 20-25 units /Kg or Alphanate or Koate DVI or Wilate
- Dose= (Patient’s weight in Kg X Desired % increase in vWF activity)/1.5
- Targets of Factor 8 activity and vWF activity:
- Major trauma, surgery, CNS bleed- 50-80%
- Delivery/ Postpartum period- >50%
- Dental extraction and minor surgeries- 30-50%
- Mucus membrane bleeds and menorrhagia- 20-80%
- Post treatment monitoring is essential, as supratherapeutic replacement doses (>200%) are associated with increased risk of thrombosis.
- Cryoprecipitates:
- Cryoprecipitates contain 5-10 times more Factor 8 and vWF than FFP
- Each cryo contains 80-100units of factor 8
- Dose in vWD is 30-60 units of Factor 8/Kg- OD
- Tranexamic acid
- Useful adjunctive therapy for both desmopressin and VWF replacement.
- Platelet transfusions
- Useful especially when haemorrhage is not controlled despite adequate factor 8 levels after Humate P
- Local measures
- Application of direct pressure
- Cold application
- Topical bovine thrombin
- Tranexamic acid
- Additional measures for menorrhagia
- OCPs
- Endometrial ablation
- Mirena insertion- Levonorgestrel releasing intra uterine device
General measures:
- Avoid aspirin and other NSAIDs
- Regular parenteral iron therapy after assessing CBC and iron profile
- Prophylaxis is usually not required as bleeding is mild and spontaneous bleeding is rare. It is useful if there is recurrent epistaxis and joint bleeds. Dose- 40-60 RCO U/kg- 1-3 times a week.
Special Situations:
- Development of inhibitors:
- Seen in case of type III disease who are multi-tranfused
- Present with loss of response to VWF concentrates
- Difficult to assess in lab
- Poor vWF recovery after factor replacement is better indicator.
- Treatment is similar to haemophilia A with inhibitors.
- Acute bleeds should be treated using recVIIa and tranexamic acid.
- Pregnancy
- In case of type 1 vWD, factor levels rise by 50%, hence no specific therapy is needed. If levels are <30%, prophylactic replacement should be done.
- In type 1 vWD- Neuraxial anaesthesia, vaginal delivery and caesarean section are safe with RICOF levels >0.5 IU/ml
- For rest of the patients (Type 2 and 3)- Factor replacement must be considered during delivery and during post-partum period. Neuraxial anaesthesia is not recommended, as restoration of normal haemostasis cannot be reliably achieved.
- Desmopressin is safe both for pregnancy and delivery, but it should be avoided if there is pre-eclampsia.
- Tranexamic acid can be given.
- Fetus may have low vWF levels, hence avoid head injury.
- Acquired Von Willebrand Disease: Refer to “acquired coagulation disorders” chapter.
Recent advances:
Recombinant von Willebrand factor and tranexamic acid for heavy menstrual bleeding in vWD
In this phase 3 crossover trial conducted at 13 haemophilia treatment centres in the USA, researchers aimed to compare the effectiveness of recombinant von Willebrand factor (VWF) with tranexamic acid in reducing heavy menstrual bleeding in female patients aged 13–45 years with mild or moderate von Willebrand disease. Participants were randomly assigned to receive two consecutive cycles of intravenous recombinant VWF or oral tranexamic acid. The primary outcome was a 40-point reduction in the pictorial blood assessment chart (PBAC) score by day 5 after two cycles of treatment. The trial was stopped early due to slow recruitment, and interim analysis data showed that neither treatment corrected the PBAC score to the normal range. Tranexamic acid was found to significantly reduce the PBAC score compared to recombinant VWF. There were no serious adverse events or treatment-related deaths.
https://doi.org/10.1016/S2352-3026(23)00119-9
An Initiative of
Veenadhare Edutech Private Limited
1299, 2nd Floor, Shanta Nivas,
Beside Hotel Swan Inn, Off J.M.Road, Shivajinagar
Pune - 411005
Maharashtra – India

howitreat.in
CIN: U85190PN2022PTC210569
Email: admin@howitreat.in
Disclaimer: Information provided on this website is only for medical education purposes and not intended as medical advice. Although authors have made every effort to provide up-to-date information, the recommendations should not be considered standard of care. Responsibility for patient care resides with the doctors on the basis of their professional license, experience, and knowledge of the individual patient. For full prescribing information, including indications, contraindications, warnings, precautions, and adverse effects, please refer to the approved product label. Neither the authors nor publisher shall be liable or responsible for any loss or adverse effects allegedly arising from any information or suggestion on this website. This website is written for use of healthcare professionals only; hence person other than healthcare workers is advised to refrain from reading the content of this website.