
howitreat.in
A user-friendly, frequently updated reference guide that aligns with international guidelines and protocols.
Essential Thrombocythemia
Updated on 12.06.2025
Introduction:
- It is a clonal myeloid disorder affecting megakaryocytic lineage characterized by sustained thrombocytosis in the blood and increased number of large mature megakaryocytes in bone marrow and clinically by episodes of thrombosis and / or haemorrhage
Epidemiology:
- Incidence: 1-2.5/ 1 lac population
- Mean age is 50- 60 Years
- Slight female predominance
Etiology: Exact cause is not known- ?Radiation ?Familial tendency
- 50-60% patients have mutation of JAK2.
- 25-35%patients have mutations in the calreticulin (CALR) gene
- 5-10% patients have MPL mutations in exon 10
- Malignant transformation occurs due to additional mutations such as TET2, ASXL1, DNMT3A, SF3B1and IDH.
- Genetic abnormalities on
- Chromosome 3 – Thrombopoietin gene
- Chromosome 1 – Thrombopoietin receptor Mpl gene
- t (5:9)- KANK1: PDGFR beta
- Over-expression of transcription factor- NF-E2
Pathogenesis:
- Genetic mutations lead to
- Increased proliferation of cells
- Cytokine hypersensitivity
- Cytokine independent inhibition of apoptosis
- Serum TPO levels are inappropriately normal or elevated.
- Platelet abnormalities in essential thrombocythemia
- Decreased number of receptors for platelet inhibitory prostaglandins like PGD2
- Increased expression of Fc receptors
- Reduction of membrane glycoprotein GpIb
- Defective coagulant activity
- Impaired serotonin binding and uptake
- Changes in platelets membrane fatty acid composition
- Abnormal arachidonic acid metabolism
- Increased glyoxalase I activity
- Increased lactate production
Clinical Features:
- Most of the patients are asymptomatic
- Excessive bruising and bleeding – Due to qualitative platelet defect and acquired vWD
- Venous / arterial thrombosis – Due to spontaneous platelet aggregation. It may result in TIA, digital infarction etc. Risk is higher with JAK2 mutation (compared to CALR) and if patient has leucocytosis.
- Microvascular disturbances: Headaches, lightheadedness, visual symptoms such as blurring and scotomata, palpitations, chest pain, erythromelalgia (Painful burning of hands accompanied by erythema due to vasoconstriction), and distal paresthesias
- Disease related fatigue and difficulties in concentration
- Splenomegaly
Disease progression:
- AML (Blast phase) or MDS: Seen in <5% cases
- Polycythaemia vera
- Myelofibrosis: Indicated by development of anemia, increased LDH and typical changes of myelofibrosis in peripheral smear. Overall risk is 10% in 15 years
Investigations:
- Hemogram:
- Marked thrombocytosis – Platelet show anisocytosis (tiny to large to giant)
- Platelet count- Usually more than 600 x 109/L
- Platelets are bizarre shaped. May have pseudopods.
- Agranular platelet cytoplasm
- Rarely leucocytosis- Neutrophilia with shift to left
- Eosinophilia& Basophilia
- Anemia is rare. RBCs – Normocytic normochromic/ microcytic hypochromic if there is chronic blood loss
- Bone marrow aspiration
- Normocellular/ hypercellular (those with mpl mutations can have hypocellular bone marrow)
- Erythropoiesis– Normal/ Increased if there is associated anemia
- Mild granulocytic proliferation
- Marked proliferation of large to giant megakaryocytes
- They are present in loose clusters
- Have abundant mature cytoplasm
- Hyperlobulated nuclei with smooth nuclear contours- "Stag horn like" nucleus
- Emperipolesis of bone marrow elements may be seen
- BM is needed prior to starting cytoreductive therapy to rule out disease progression to MF
- Bone marrow biopsy:
- Megakaryocytes form clusters within interstium
- Reticulin fibres – Normal / mildly increased
- Following features indicate alternate diagnosis (PV, prefibrotic PMF, PMF or a form of MDN/MPN)
- Marked hypercellularity
- Atypical megakaryocytes with hyperchromatic nuclei
- Tight clustering of the megakaryocytes
- Dysplastic feature in any of the lineages
- Significant increase in reticulin (grade 2 or more on a scale of 0-3)
- Immunophenotyping- Nothing specific
- Cytogenetics
- Nothing specific (No Ph Chromosome)
- Karyotyping abnormality is present in 5-10% of patient
- Abnormalities include– del (13q22), +8, +9
- Bleeding time – prolonged
- Serum cobalamin and unsaturated cobalamin binding capacity – Raised
- Serum uric acid, LDH and acid phosphatase levels- Raised (Due to increased cell turnover)
- Serum K+ - Raised, known as pseudohyperkalemia. (Occurs due to in vitro release of K+ from platelets)
- Tests for haemostasis (PT & APTT) – Normal
- Platelet function test- Defective aggregation with epinephrine
- MPN mutation panel including BCR-ABL to exclude CML.
Criteria for Diagnosis: Either all major criteria or the first 3 major criteria plus, the minor criterion should be met.
- Major criteria
- Platelet count >4,50,000/cmm
- Bone marrow biopsy showing proliferation mainly of the megakaryocytic lineage, with increased numbers of enlarged, mature megakaryocytes with hyperlobulated nuclei; no significant increase or left shift in neutrophilgranulopoiesis or erythropoiesis; rarely a minor (grade 1) increase in reticulin fibres
- WHO criteria for CML, MF, PV or other myeloid neoplasms( such as MDS with del5(q), refractory anaemia with ringed sideroblasts with thrombocytosis) are not met
- Positive for JAK2, CALR, or MPL mutation
- Minor criterion
- Presence of a clonal marker or absence of evidence of reactive thrombocytosis (Underlying inflammation or infection/ Underlying neoplasm/ Prior splenectomy)
Criteria for diagnosis of Post ET myelofibrosis:
- Required criteria (Both are required)
- Documentation of a previous diagnosis of ET as defined by WHO criteria
- BM fibrosis of grade 2-3 (On a scale of 0-3)
- Additional criteria (2 are required)
- Anaemia and 2gm/dL fall of haemoglobin from baseline
- Leucoerythroblastic blood film
- Increasing splenomegaly
- Elevated LDH
- Any one constitutional symptom- More than 10% weight loss in 6 months, night sweats, unexplained fever
Progression of disease:
- ET- Accelerated phase: BM/PB- Blasts- 10-19%
- ET- Blast crisis: BM/PB- Blasts ≥19%
Prognosis:
- Median survival: 10-15 years (Life expectancy is near normal for most of the patients)
- Splenectomy is associated with worst prognosis (As it causes dramatic increase in platelet count)
- The triple A survival risk model:
- Age >70 years (4 points)
- Age 50–70 years (2 points)
- Absolute neutrophil count: ≥8000/cmm (1 point)
- Absolute lymphocyte count: <1,700/cmm (1 point)
| Points | Risk category | Median survival (in years) |
| 5-6 | High risk | 8 |
| 4 | Intermediate risk-1 | 14 |
| 2-3 | Intermediate risk-2 | 21 |
| 0-1 | Low-risk | 47 |
- Mutation-enhanced International Prognostic Scoring System for ET (MIPSSET)
- Presence of adverse mutations (SRSF2, SF3B1, U2AF1 and TP53): 2 points
- Age >60 years: 4 points
- Male: 1 point
- Leukocyte count ≥11,000/cmm: 1 point
Points | Risk | Median survival (in years) |
≥ 4 | High-risk | 3.2 |
2-3 points | Intermediate-risk | 13.1 |
0-1 | Low-risk | 24 |
Risk factors for transformation to post-ET myelofibrosis:
- Age ≥60 y
- Bone marrow fibrosis grade >1
- Anaemia
- Leukocytosis
- JAK2 p.V617F negative disease
- MPL and CALR-1 mutations
- Mutations in epigenetic regulators: ASXL1
- Therapy resistance specifically hydroxycarbamide
- Certain additional mutations: SH2B3, SF3B1, U2AF1, TP53, IDH2, and EZH2.
Risk factors for transformation to blast crisis:
- Age ≥60 y
- Leukocytosis
- Abnormal karyotype
- Thrombosis
- Platelets >10 lacs/cmm
- Reticulin fibrosis
- Therapy resistance specifically hydroxycarbamide
- Leukemogenic therapy (radiophosphorous, alkylators)
- Additional mutations: RUNX1, SH2B3, SF3B1, U2AF1, TP53, IDH2, and EZH2
Differential Diagnosis:
- Other causes of thrombocytosis: Refer Thrombocytosis section (Click Here)
Pretreatment Work-up:
- History: Thrombotic/ Cardiovascular risk
- Examination:
- Spleen (cm):
- BMA and Bx
- Haemoglobin
- TLC, DLC
- Platelet count
- Peripheral smear
- LFT- Bili- T/D SGPT: SGOT: Albumin: Globulin:
- Creatinine
- Uric acid
- LDH
- HIV
- HBsAg
- HCV
- UPT
- CRP
- Iron studies
- Cytogenetics
- BCR-ABL1
- JAK 2
- CAL-R
- MPN
- Chemotherapy consent after informing about disease, prognosis, cost of therapy, side effects, hygiene, food and contraception
- Tumor board meeting and decision
- Inform primary care physician
Treatment Plan:
Risk stratification (Modified IPSET thrombosis score):
- Very low (Survial: Same as normal population)
- Age <60 years
- No JAK2 mutation
- No prior history of thrombosis
- Low risk (Survial: Same as normal population)
- Age <60 years
- JAK 2 mutation- Present
- No prior history of thrombosis
- Intermediate risk (Survial: 24.5 years)
- Age >60 years
- No JAK 2 mutation
- No prior history of thrombosis
- High risk (Survial: 14.7 years): Any one
- History of thrombosis at any age
- Age >60 years with JAK2 mutation/ significant thrombotic risk factor such as diabetes / hypertension
- Platelet count > 1500 x 109 /L
For all patients
- Manage cardiovascular risk factors
- Stop smoking
- Control of hypertension
- Early detection and treatment of diabetes
- Treatment of dyslipidemia
- Aspirin
- 81-100mg-OD
- To be given to all patients
- Avoid in patients with extreme thrombocytosis with possibility of acquired von Willebrand disease
- May not be useful in CALR mutated ET and has higher risk of bleeding in them.
- Twice daily may be considered for patients with refractory symptoms or those with arterial thrombosis.
- Avoid giving it along with Clopidogrel, as it increases the risk of bleeding
For Very low/ Low/ Intermediate risk patients: Monitor every 2-3 monthly for indications to start cytoreductive therapy such as
- New thrombosis
- Acquired vWD
- Disease related major bleeding
- Splenomegaly
- Progressive thrombocytosis or leucocytosis
- Disease related symptoms such as pruritus, weight loss or night sweats
- Vasomotor/ microvascular disturbances (headaches/ chest pain, erythromelalgia) not responsive to aspirin
High Risk patients
(and Very low/ Low/ Intermediate risk patients with indications for cytoreduction)
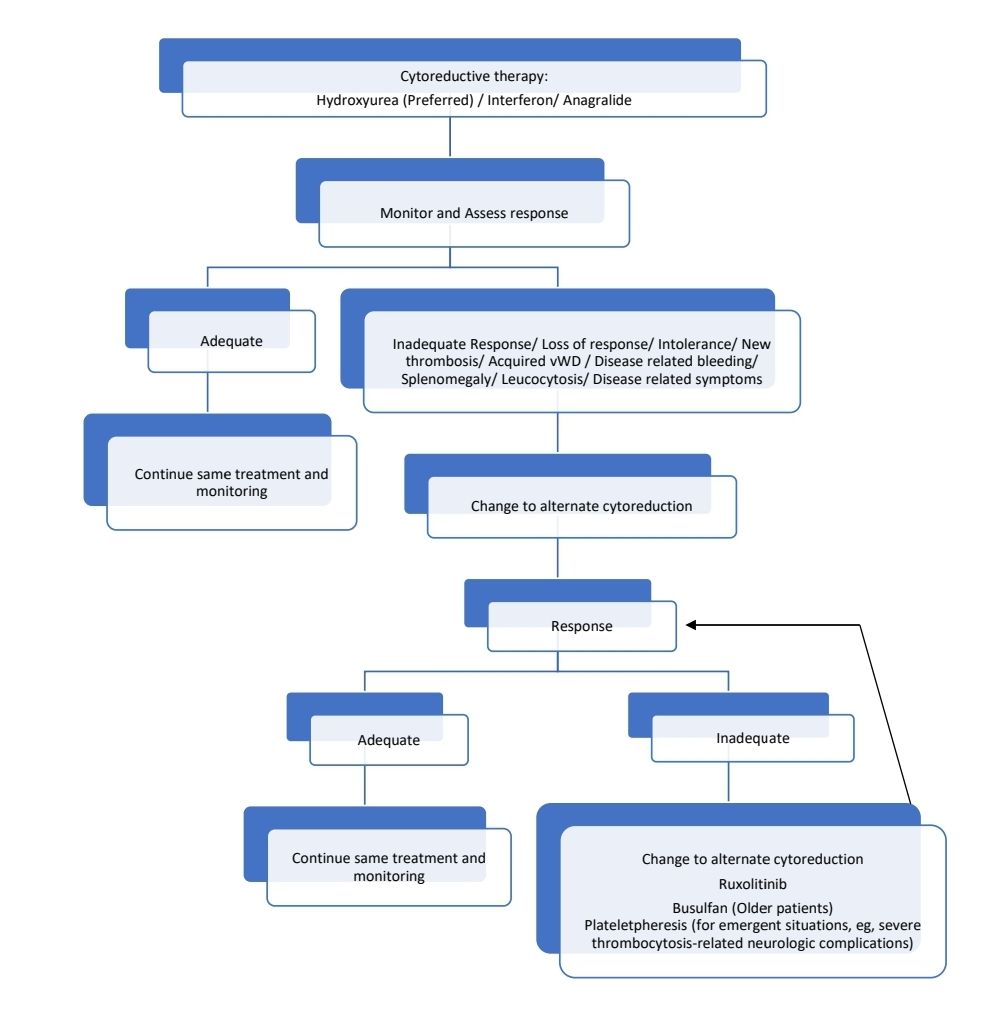
Goal: To keep platelet count <6lacs/cmm (<4lacs/cmm if possible) and limit thrombo-hemorrhagic risk.
Progression of disease:
- Myelofibrosis: Treat similar to primary myelofibrosis
- AML:
- Fit: Induction followed by AlloSCT
- Frail: Hypomethylating agents
Response Criteria:
- Complete remission:
- Durable (>12 weeks) resolution of disease related symptoms and signs including splenomegaly
- Durable peripheral blood count remission (Platelet count <4lac/cmm, TLC <10,000/cmm, and absence of leucoerythroblastosis)
- No signs of progressive disease
- Absence of thrombotic or haemorrhagic events
- Bone marrow- Disappearance of megakaryocytic hyperplasia with <2 grade fibrosis
About Each Modality of Treatment:
- Hydroxyurea
- Dose- 500-2000mg/day
- Defining criteria for resistance or intolerance to hydroxyurea
- Platelet count of >6lac/cmm, after 3 months of at least 2gm/day of hydroxyurea
- Platelet count of >4lac/cmm, with WBC count <2500/cmm or Haemoglobin<10gm/dL at any dose of hydroxyurea
- Presence of leg ulcers or other unacceptable mucocutaneous manifestations at any dose
- Hydroxyurea related fever
- Risk of leukaemia is not high compared to untreated patient
- Interferon- alfa
- Dose- 3 million units-OD
- Preferred choice in patients aged less than 40 years.
- Useful especially in pregnancy
- Pegylated interferon: 45-90microgm-SC- weekly once. Titrate up to 180 mcg once-a-week
- Ropeginterferon: 100 mcg every 2 weeks. Titrate up to 300 mcg
- Suppresses pluripotent and lineage committed progenitors
- Side effects: Depression, fatigue, hepatitis, pneumonitis
- Mean time for response- 3 months
- Anagrelide:
- Dose: 0.5 mg PO- BD- Slowly increase to QID. May increase dose based on platelet count, which should be done every week. Do not exceed dose of 10mg/day
- Median response time- 2.5-4 weeks.
- Inhibits cyclin nucleotide phosphodiesterase III and phospholipase A2, which leads to inhibition of megakaryocyte differentiation.
- Side effects include: Vasodilation, reduced platelet aggregation, diarrhoea, anaemia, palpitations and arrythmias, fluid retention, heart failure and head ache
- Useful as second line therapy
- There is increased risk of myelofibrotic transformation. Hence bone marrow biopsy is needed every 3 years.
- Busulfan
- Dose- 60microgm/kg (Max- 4mg)
- Continue till platelet counts are less than 4lac/cmm. Then stop. Restart when there is rise in platelet count again.
- Can cause pulmonary toxicity when cumulative dose is between 500-5700mg.
- Long term use can cause leukaemia
- Used in patients with limited life expectancy
- Ruxolitinib:
- Decreases thrombocytosis, ET- related symptoms and splenomegaly.
- Radioactive phosphorous (32p)
- 150-300 MBq- IV.
- Can be repeated after 3 months.
- Useful in elderly patients with limited life expectancy.
- Other drugs which are under trial:
- Pelabresib (Bromodomain inhibitor)
- Bomedemstat (LSD1 inhibitor)
- INCA033989 (Monoclonal antibody targeting mutant CALR)
Supportive Care:
- Thrombosis
- Initial therapy as per ACCP guidelines
- If thrombosis is life threatening, immediate platelet pheresis with a target to decrease platelet count to less than 5lac/cmm, then start cytoreductive therapy
- No clear data on duration of anticoagulation.
- DOACs can be used safely
- If there is high risk of recurrence or if thrombotic event was life threatening, better to continue anticoagulation lifelong.
- Combined OC pills and HRT are discouraged in women with ET.
- Ovarian stimulation therapy is associated with high risk of thrombosis
- For Pruritus-Cimetidine
- Bleeding- Paradoxically need platelet transfusions.
Special Situations:
- Essential thrombocythemia and pregnancy
- ET is most common MPN seen at child bearing age group
- In pregnancy most common complication of ET is first trimester miscarriage (30% of pregnancies) - occurs due to placental infarcts
- Other complications include intra uterine death, growth retardation, premature delivery, pre eclampsia, maternal thrombosis and haemorrhage
- Treatment:
- Low risk – Aspirin alone
- High risk (Prior h/o thrombosis / Platelet count of >10lac /cmm/ Prior pregnancy complications/ Severe preeclampsia)
- IFN alfa + Aspirin
- Hydroxyurea &Anagrelide should not be used because of their teratogenic effect
- Regular maternal and foetal monitoring
- Uterine artery doppler scan at 20-24 weeks
- Stop aspirin 5 days prior to delivery
- Avoid dehydration during labour
- Thromboprophylaxis with LMWH after delivery, after complete haemostasis is achieved.
- Continue LMWH for 6 weeks after delivery
- Breast feeding is safe with LMWH
- ET in children
- Diagnosis is made after all causes of thrombocytosis are ruled out
- ET is extremely rare in children and raised platelet count is often due to "physiological recalibration" of platelet count to higher level.
- Closely monitor these children, as they may transform in to AML or myelofibrosis
- Aspirin must be used with caution, due to risk of Reye's syndrome.
- Screen for JAK2 and MPL mutations
- If indicated use Hydroxyurea- 15mg/kg/day for decreasing platelet counts.
- Surgery in ET
- Stop aspirin 7-10 days prior to surgery
- 5-fold higher risk of VTE
- Post-operative thromboprophylaxis with LMWH must be given.
- Try to decrease platelet count to <4lac/cmm prior to surgery
- Post ET blast transformation
- Has extreme poor prognosis (98% mortality within 3 months regardless of any therapy)
- Median survival- 5 months
- May respond to demethylating agents. Once in CR, better survival with allogeneic stem cell transplant.
Related Disorders:
- Acquired sideroblastic anaemia associated with thrombocytosis
- Features of essential thrombocytosis, associated with ringed sideroblasts in marrow
- Associated with JAK2 mutation
- Has good prognosis
Figures:
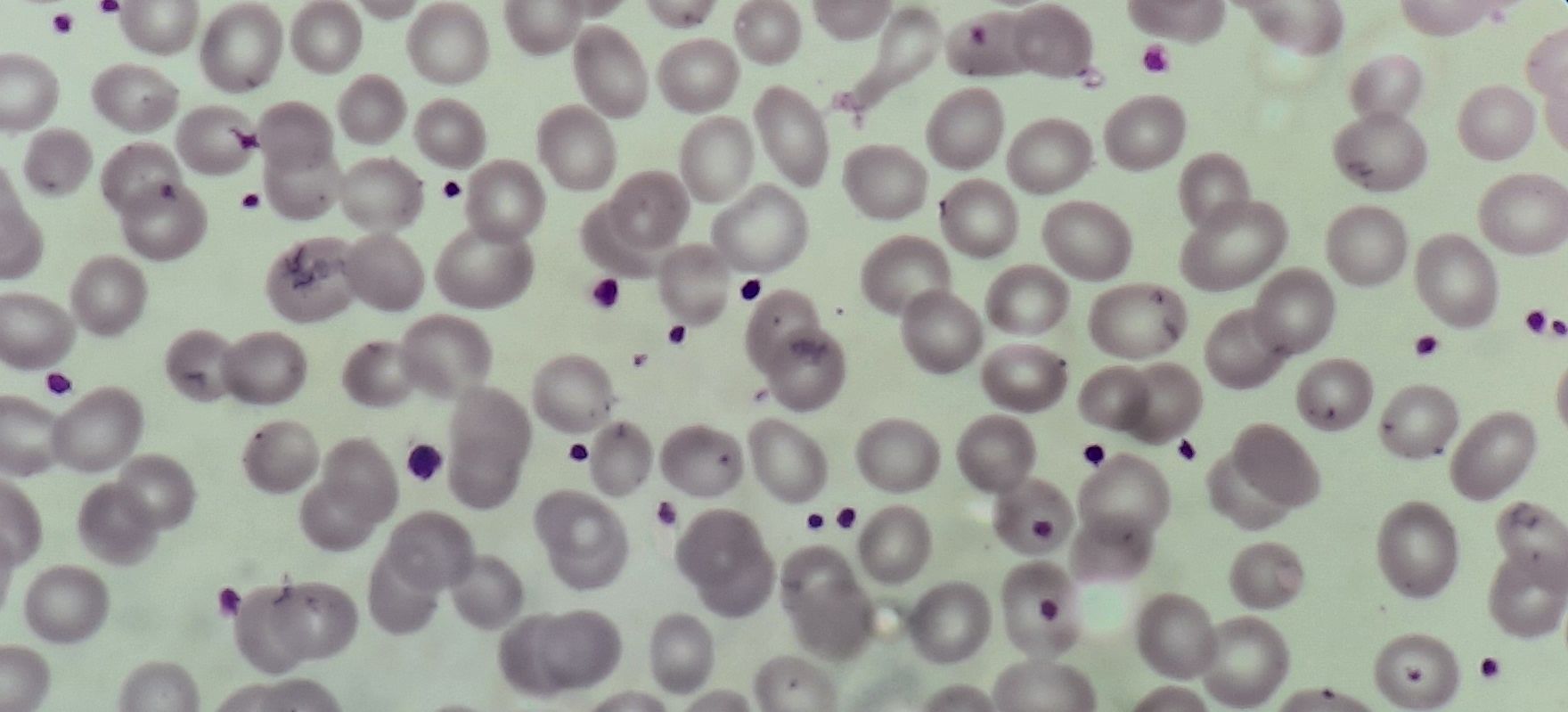
Essential thrombocythemia- Peripheral smear
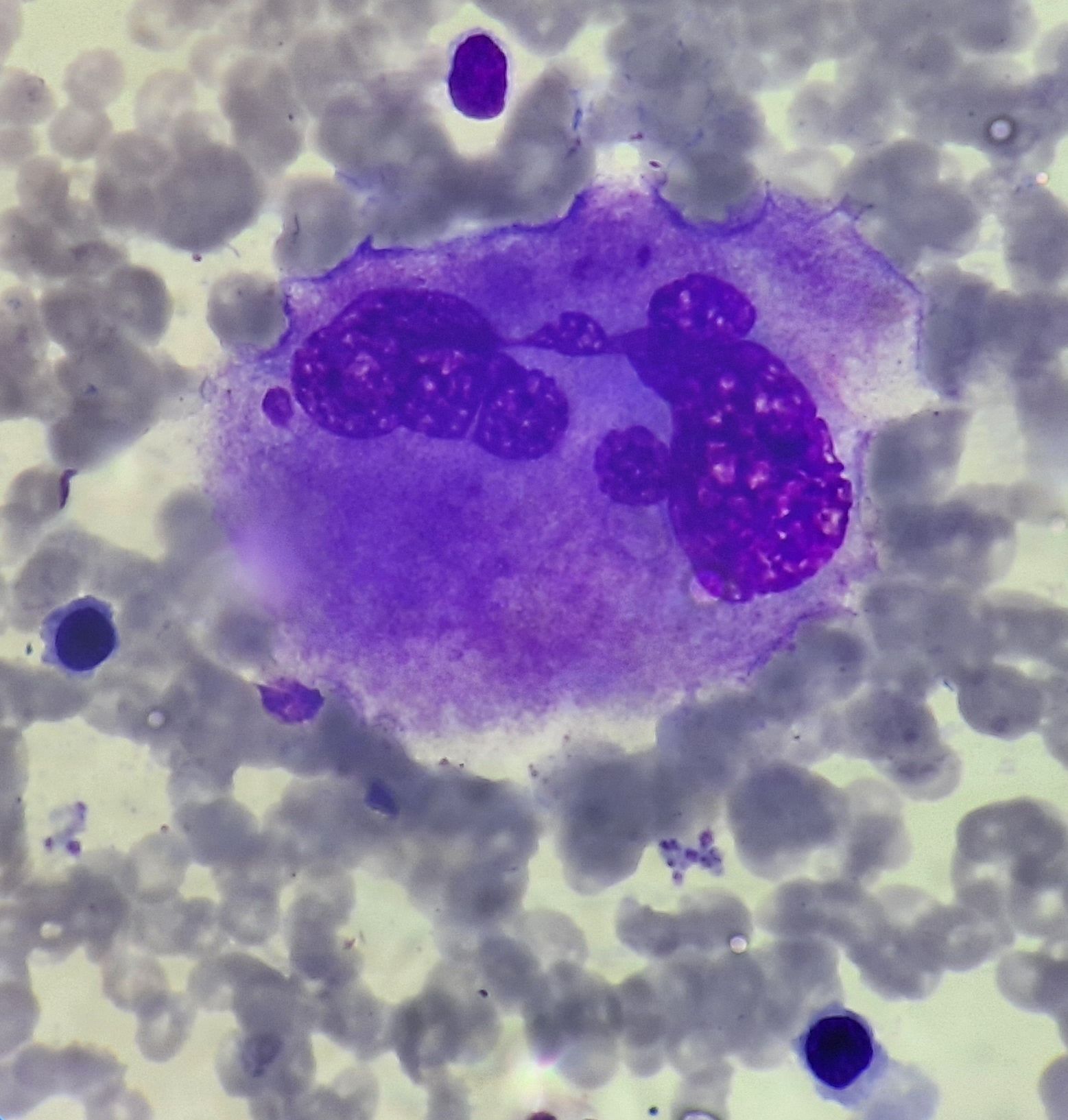
Essential Thrombocythemia- Megakaryocyte
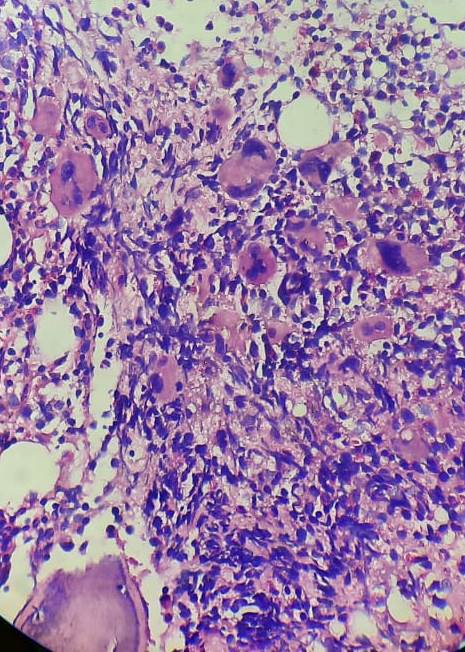
Essential Thrombocythemia- Bone marrow biopsy
Recent advances:
Cytoreductive treatment and association with platelet function and maturity in patients with essential thrombocythaemia
Present study investigated the effect of cytoreductive treatment on platelet function and turnover in ET patients. Patients not receiving cytoreductive treatment had significantly higher platelet aggregation 24 h after aspirin intake than the other ET groups. Patients receiving hydroxycarbamide had significantly higher expression of platelet granule makers, P-selectin and CD63, than patients receiving peg-IFN. Cytoreduction provides more consistent platelet inhibition compared with no cytoreductive treatment. Moreover, peg-IFN provides superior inhibition of platelet activation markers than hydroxycarbamide.
https://doi.org/10.1111/bjh.18303
Thrombotic risk factors in patients with essential thrombocythaemia
The 2016 revised International Prognostic Score for Thrombosis in Essential Thrombocythaemia-thrombosis (r-IPSET-t) score stratifies patients into very-low- (VLR), low- (LR), intermediate- (IR) and high-risk (HR) groups. Present study validated the r-IPSET-t in the biggest population of patients with ET (n = 1381). With an average follow-up of 87.7 months, there were 0.578 thrombotic events/person-year and 0.286 bleeding events/person-year after diagnosis. The 10-year thrombosis-free survival was 88% and 99% for the r-IPSET-t LR and VLR groups. Cytoreduction was a thrombotic risk factor in younger patients (aged <60 years. The European LeukemiaNET (ELN) does not recommend aspirin for VLR patients but in this real-life analysis 83.1% of VLR patients received it.
https://doi.org/10.1111/bjh.18387
Impact of non-driver gene mutations on thrombo-haemorrhagic events in ET patients
This study, involving 579 essential thrombocythemia (ET) patients, analyzed the association between non-driver gene mutations and thrombo-hemorrhagic events. ASXL1 and TP53 mutations were linked to thrombosis, while DNMT3A mutations were associated with hemorrhage. Patients with ASXL1 or TP53 mutations had worse thrombosis-free survival rates, and JAK2V617F-mutated patients with ASXL1 mutations had shorter survival. DNMT3A-mutated patients showed shorter hemorrhage-free survival. The findings suggest that considering non-driver gene mutations is crucial for personalized treatment strategies in ET.
https://doi.org/10.1111/bjh.19177
Aspirin in essential thrombocythemia: The ARES trial
In this randomized phase-2 trial, 242 patients with essential thrombocythemia (ET) were compared between twice-daily and once-daily low-dose aspirin regimens over 20 months. Twice-daily aspirin resulted in better suppression of thromboxane biosynthesis (TXB2) and reduced disease symptoms, including microvascular pain. While clinically relevant non-major bleeding was slightly higher in the twice-daily group, there were fewer major thromboses. There was no significant difference in major bleeding or gastrointestinal discomfort between the two regimens, suggesting twice-daily aspirin may offer better long-term efficacy without compromising safety.
https://doi.org/10.1002/ajh.27418
An Initiative of
Veenadhare Edutech Private Limited
1299, 2nd Floor, Shanta Nivas,
Beside Hotel Swan Inn, Off J.M.Road, Shivajinagar
Pune - 411005
Maharashtra – India

howitreat.in
CIN: U85190PN2022PTC210569
Email: admin@howitreat.in
Disclaimer: Information provided on this website is only for medical education purposes and not intended as medical advice. Although authors have made every effort to provide up-to-date information, the recommendations should not be considered standard of care. Responsibility for patient care resides with the doctors on the basis of their professional license, experience, and knowledge of the individual patient. For full prescribing information, including indications, contraindications, warnings, precautions, and adverse effects, please refer to the approved product label. Neither the authors nor publisher shall be liable or responsible for any loss or adverse effects allegedly arising from any information or suggestion on this website. This website is written for use of healthcare professionals only; hence person other than healthcare workers is advised to refrain from reading the content of this website.