
howitreat.in
A user-friendly, frequently updated reference guide that aligns with international guidelines and protocols.
Hereditary Spherocytosis and Other Membrane Abnormalities
Updated on: 10.05.25
Classification of protein abnormalities in the erythrocyte membrane that result in vertical or horizontal interaction defects and cause hemolytic anemia
- Vertical interaction defects (Produces spherocytic anemia)
- Primary spectrin deficiency
- Secondary spectrin deficiency due to defects or deficiencies in Protein 4.2, Ankyrin and Band 3
- Horizontal interaction defects (Produce elliptocytosis & other bizarre forms)
- Actin
- Protein 4.1
- Adductin
Detailed structure of RBC membrane is available at: https://howitreat.in/hematology/red-blood-cell
Hereditary Spherocytosis
Introduction:
- Hereditary spherocytosis is an autosomal dominant condition, occurring due to defect in vertical interaction, characterized by abnormal shape and flexibility of RBCs.
Epidemiology:
- Highest prevalence is seen in north European population (1 in 2000 births)
Etiology:
- Mutation of genes encoding:
- Ankyrin- ANK 1
- Band 3- SLC4A1
- Beta spectrin- SPTB1
- Biallelic defects in band 4.2- EPB42
- Biallelic defects in Alpha spectrin- SPTA1
Pathogenesis:
Deficiency of spectrin, ankyrin, band 3, protein 4.2 etc.
↓
Weakening of the vertical interaction
↓
Reduced membrane stability and spontaneous loss of cell membrane due to rapid vesciculation (process by which small, membrane-bound sacs i.e. vesicles are formed and bud off from the cell membrane)
↓
Reduction of cell surface to volume ratio
(Surface becomes less while volume remains same)
↓
RBC is forced to assume smallest possible diameter for a given volume i.e. sphere.
↓
As spherocytes have reduced flexibility, they are trapped in splenic sinusoids
↓
Extravascular hemolysis
Note:
- Complete absence of spectrin is not viable, so homozygous children are not found.
- Clinical severity depends on degree of spectrin deficiency
Classification:
| Trait/ Carrier | Mild | Moderate | Severe |
Hemoglobin (gm/dL) | Normal | 11-15 | 8-12 | 6-8 |
Reticulocyte % | Normal | 3-6 | >6 | >10 |
Bilirubin | <1.7 | 1.7-3.4 | >3.4 | >5.1 |
Spectrin molecules per RBC | 100 | 80-100 | 50-80 | 40-60 |
Splenectomy | Not required | Usually not required | Necessary during school age, before puberty | Necessary at early age (Delay until 6 years if possible) |
Clinical Features:
- Varying presentation- Asymptomatic (compensated hemolysis) to marked hemolysis leading to hydrops fetalis. Severity depends on extent of loss of spectrin. Severity is relatively uniform in a given family.
- Anemia- usually 5-30 days after birth as functioning of spleen becomes mature only after birth. But diagnosis may be made at any age depending on severity.
- Splenomegaly- mild to moderate, may manifest with early satiety or left upper quadrant fullness.
- Gall stones- Pigmented type
- Acholuric jaundice- Jaundice with absence of bile in urine. Usually present since birth.
- Family history: Found in 75% cases
- 3 types of crisis may be seen in hereditary spherocytosis
- Hyperhemolytic crisis
- Triggered by infection
- Presents with fever, abdominal pain, splenic enlargement, Jaundice, hypotension and shock
- Aplastic crisis
- Triggered by parvovirus, trauma, surgery, pregnancy
- Presents as sudden worsening anemia
- Megaloblastic crisis
- Occurs due to acquired folic acid deficiency, usually in high demand situations such as pregnancy.
- Hyperhemolytic crisis
- Skin lesions- Leg ulcers, gouty tophi, chronic leg dermatitis, (related to decreased RBC deformability)
- Extramedullary erythropoiesis- Masses may form in spine, kidney etc
- Poor growth, deformities of hands and skull
- Predisposition to malignancy- MPN, myeloma etc
- Iron overload- May be seen even in untransfused patients due to associated HFE mutation (Ferritin >500mg/l and transferin saturation of >60% indicates iron overload)
- Angioid streaks in optic fundi
- Venous thromboembolism and pulmonary hypertension
- Rare non-hematological manifestations:
- Neuromuscular abnormalities (as ankyrin and spectrin are expressed in neural tissues)-slowly progressive spinocerebellar degenerative disease, spinal cord dysfunction, movement disorders, cardiomyopathy
- Distal renal tubular acidosis is seen with defects with band 3.
Investigations:
(Findings are not very typical in newborns except high MCHC, hence better to rescheduled until the sixth month of age or later)
- Hemogram:
- Hemoglobin content- Normal/decreased
- High quality smears are needed as spherocytes may appear as artifacts
- Spherocytes- Abnormally small in sized RBCs which have uniform shape and lack central 1/3rd of pallor
- Polychromatophilic cells and nucleated RBCs may be seen.
- Pincered erythrocytes are seen in Band 3 deficiency.
- Spherocytic acanthocytes are seen in beta spectrin deficiency
- Spherocytes and stomatocytes may be seen in Japanese protein 4.2 deficiency
- MCV- Normal/decreased
- MCH – Normal
- MCHC – Increased due to cellular dehydration (>36g/dL)- It is the only anemia where MCHC is increased. Laser based counters provide histograms with hyperdense erythrocytes (MCHC >40) which detects all cases of HS. Hence it is a best screening tool.
- RDW- <14%
- HS ratio- MCHC/MCV- If more than 0.36, it indicates presence of HS.
- WBC and platelets – Normal
- Reticulocyte count- >8%
- Mean reticulocyte volume: Decreased
- Reticulocyte count/ immature reticulocyte fraction ratio: Mild cases: More than 19 and severe cases More than 7.7.
- Osmotic fragility test
- Increased fragility
- Hemolysis is complete between 0.5-0.4% NaCl
- Normal test does not rule out HS.
- False negative test is found in newborns due to the high level of fetal hemoglobin
- Positive test is also seen in hereditary elliptocytosis, and autoimmune hemolytic anemia
- Osmotic gradient ektacytometry- Measures deformability of whole RBC
- Serum bilirubin-Raised (Mostly indirect)
- Fecal urobilinogen- Raised
- LDH – Raised
- Haptoglobin-Decreased
- DCT and ICT- Negative (needed, as spherocytes are seen in AIHA as well)
- Peripheral smear of family members for HS
- Autohemolysis test
- It has value in differentiating various types of congenital nonspherocytic hemolytic anemias
- It measures the degree of spontaneous hemolysis of blood incubated at 37o C
- Degree of hemolysis depends on integrity of cell membrane & adequacy of cell enzymes involved in glycolysis.
- Normal – 0.2 – 2% hemolysis after 24- 48 hours.
- Abnormalities
Type | Observation | Seen in |
Type I | 2-6% auto hemolysis, but significant correction when glucose is added | PNH and G6PD deficiency |
Type II | 8-44% hemolysis& glucose has no effect | Pyruvate kinase deficiency |
Type – III | 5-25% hemolysis at 24 hours & 75% at 48 hrs If glucose is added before incubation hemolysis is significantly reduced | Hereditory spherocytosis |
- Cryohemolysis test:
- HS RBCs have a high susceptibility for lysis when rapidly cooled from 37 degree C to 0 degree celcius.
- Acidified glycerol lysis test
- Glycerol slows down entry of water into cells in vitro.
- The time taken for lysis to occur is a function of the osmotic resistance of the cells
- Hereditory spherocytosis cells lyses more rapidly than normal cells
- Test is easier to perform than osmotic fragility test.
- Sodium dodecylsulphate solubilized polyacrylamide gel (SDS-PAGE) electrophoresis
- Used to quantify proteins in the membrane of RBCs
- This is the confirmatory test for HS.
- Results are expressed as ratios of individual RBC membrane proteins to Band 3.
- Done only in specialized diagnostic centres and is time-consuming
- Flow cytometry:
- Binding of eosin labeled maleimide (EMA binding test)
- Cells deficient in band 3 fail to bind to EMA
- Most accurate screening test, with a relatively short time-around-time, high sensitivity, economical, and feasible. It can be done even in patients who have been transfused.
- Other conditions giving reduced fluorescence: South east asian ovalocytosis, Congenital dyserythropoietic anemia Type-II, Hereditary pyropoikilocytosis and Cryohydrocytosis
- Interpretation (Based on grade of fluorescence):
- Positive for HS- Fluorescence is > 21%
- Negative- Fluorescence is < 16%
- Equivocal- If between 16% and 21%
- Identification of gene defects by NGS-
- Involves sequencing of relevant genes to identify mutations in them.
- Widely available, cost effective and highly reliable.
Criteria for diagnosis:
- History (including family history) and clinical findings (splenomegaly) suggestive of HS.
- Presence of spherocytes and reticulocytosis
- Negative direct antiglobulin test
- If diagnosis is equivocal/ otherwise also: Preferable to do genetic testing (by NGS) for specific genetic mutations.
Treatment:
- Folic acid supplementation- Life long for moderate and severe HS.
- Transfuse PRBCs as and when required
- Severe neonatal jaundice:
- Phototherapy +/- exchange transfusion
- Some children may need transfusion support for almost a year due to inability to mount an adequate erythropoietic response. Erythropoietin may be used until age of 9 months.
- Splenectomy:
- Restores the life-span of red cells to normal and hence cures hemolysis & hyperbilirubinemia.
- Should be done after 6 years of age except in patients with severe anemia
- Dehydrated hereditary stomatocytosis is a contraindication for splenectomy, hence this must be ruled out.
- Indications:
- Marked hemolysis requiring multiple transfusions (Severe HS)
- Growth retardation/ skeletal changes
- Anemia induced end organ damage
- Development of leg ulcers
- Appearance of extramedullary hematopoietic tumors
- Laporoscopic is more feasible and safe. Any splenunculi (accessory splenic tissue) must be removed to prevent recurrence of symptoms.
- Routine postoperative thromboprophylaxis should be given for adults.
- Vaccinations (Pneumococcus, Hemophiluc influenza type b and meningococcus) must be given before procedure.
- Prophylactic penicillin must be given for several years.
- Cholecystectomy may be performed at the same time, if there is cholelithiasis (even if asymptomatic).
- Chelation therapy/ phlebotomies- if there is iron overload.
- Counseling to potential parents regarding possibility of neonatal jaundice and need for exchange transfusion
Hereditary Elliptocytosis
(Hereditary Ovalocytosis)
- Autosomal dominant condition
- Gene affected is closely related to Rh locus on chromosome 1
- Classification
Type of HE | Hemolysis | Erythrocyte Shape |
| Common HE | Variable; minimal to severe | Elliptocytes |
Spherocytic HE (hemolytic ovalocytosis) | Present | Spherocytes and fat elliptocytes |
Southeast Asian ovalocytosis (Stomatocytic HE Melanesian/ South east Asian ovalocytosis) | Mild or absent | Roundish elliptocytes that are also stomatocytic (Spoon shaped RBCs)- Oval cells with 1-2 longitudinal slits |
- Defect involves horizontal membrane protein interactions
- Actin
- Protein 4.1
- Adductin
- Glycophorin C
- RBCs fail to recover their biconcave shape after they acquire elliptical shape in microcirculation where they are subjected to shear stress.
Clinical features
- 90% do not have hemolysis
- Asymptomatic HE patients may experience hemolysis in association with infections, hypersplenism, Vitamin B12 deficiency or microangiopathic disorders.
- Stomatocytic HE Melanesian/ South east Asian ovalocytosis children can present with neonatal jaundice, but hemolysis resolves by 3 years of age. Homozygous status results in hydrops fetalis.
- Some can have chronic hemolysis
Investigations:
- Hemogram
- Hemoglobin: Usually >12g/dL
- Prominent elliptocytosis: >25% RBCs & usually >60% (<25% are seen with megaloblastic anemia and iron deficiency anemia)
- Pseudoelliptocytes (Artifacts during smear preparation)- They are seen only near the tail. Long axes of all pseudo elliptocytes are parallel.
- Reticulocyte count – Mild elevation – >4%
- In hemolytic hereditary elliptocytosis, lab findings due to hemolysis are seen
- Osmotic fragility test- Abnormal only in severe HE
Treatment
- Splenectomy for hemolytic variant (Otherwise no treatment is needed)
Note: Elliptocytes are resistant to malarial infection due to abnormal rigidity of membrane
Hereditary Pyropoikilocytosis
- Autosomal recessive
- Presents in infancy as severe hemolytic anemia with extreme poikilocytosis
- Cells fragment when heated to 45o-46 o C (Normal – 50 o C)
- They disintegrate when incubated at 37 o C for more than 6 hours.
- Pathogenesis:
Mutant spectrin- Prevents self association of heterodimers of spectrins
↓
Disruption of membrane skeletal lattice & Membrane cell destabilization
↓
Erythrocyte fragmentation & poikilocytosis
- Peripheral smear: Morphologic abnormalities of RBCs- Budding, fragments, microspherocytes, elliptocytestriangulocytes.
- MCV – Decreased (25-55fL)
- Auto hemolysis- Increased and not corrected by glucose.
Hereditary Stomatocytosis
- Autosomal dominant condition
- 4 variants depending on intracellular sodium concentration (Normal- 5-10 mmol/l)
- Dehydrated Hst- MCHC increased- Na- 12-18mmol/L
- Overhydrated Hst- MCHC decreased- Na- >60mmol/L
- Cryohydrocytosis (Temperature sensitive leak) - Na- 20-50 mmol/L
- Pseudohyperkalemia- Na- Normal
- Normal:
Shear stress and pressure on RBC in narrow capillaries
↓
Activation of PIEZO1
↓
Influx of Ca 2+ into the cytoplasm of RBC
↓
Activation of KCNN4
↓
Increased efflux of K+ out of RBC
↓
Drawing of water out of RBC
↓
Reduced volume of RBC
↓
Easy passage through capillary which is 1/3rd of RBC size
- In dehydrated hereditary stomatocytosis:
Mutation in PIEZO1 and KCNN4 genes
↓
Net loss of intracellular K+ exceeds the passive Na+ influx
↓
Net Na+ gain
↓
Dehydration of cells
↓
Intravascular hemolysis
- In overhydrated hereditary stomatocytosis:
Defect in stomatin gene
↓
Deficiency of band 7 protein (Abnormality in RBC lipids)
↓
RBC membrane is abnormally permeable to both Na+ & K+
↓
Net gain of sodium is greater than net loss of potassium
↓
Over hydration of cells
↓
Hydrocytes look like stomatocytes in dried a stained blood films
(Slit mouth like area of pallor)
- Clinical features:
- Compensated hemolytic anemia- Jaundice, splenomegaly, gall stones
- Sometimes- Recurrent fetal losses, hydrops fetalis, neonatal hepatitis, familial pseudo-hyperkalemia
- Investigations:
- Wet smear – Appear uniconcave & bowl shaped.
- Peripheral smear shows stomatocytes (10% to 50%), macrocytosis
- Osmotic fragility- Increased
- Autohemolysis-Increased (Partially corrected by ATP & glucose)
- Treatment:
- Folate supplements
- Splenectomy in selected cases- Should not be done in dehydrated variant as it is associated with marked thrombotic tendencies.
- Acquired causes of stomatocytosis
- Acute alcoholism
- Liver disease
- Cardiovascular disease
- Artifacts in peripheral smear
- Syndromic forms include:
- Stomatin deficient cryohydrocytosis with mental retardation, seizures, spastic rigidity, ataxia and hepatosplenomegaly (Mutation in SLC2A1 gene)
- Phytosterolemia nonleaky stomatocytosis with macrothrombocytopenia (Mutation in ABCG5 or ABCG8 genes): Discussed below
Sitosterolemia (Phytosterolemia)
- Autosomal recessive
- Mutations in genes encoding transporter proteins- ABCG5 or ABCG8
- Pathogenesis:
Increased absorption and decreased excretion of sterols
↓
Plasma levels of plant sterols is increased
↓
Accumulation of plant sterols in cell membranes
↓
Hemolytic anemia
- Diagnosis confirmed by genetic tests
- Clinical features:
- Extreme phenotypic heterogeneity
- Xanthomatosis with severe hypercholesterolemia
- Early onset premature cardiovascular disease due to accelerated atherosclerosis
- Hemolytic anemia with stomatocytosis, macrothrombocytopenia, splenomegaly and abnormal bleeding
- Splenomegaly
- Treatment
- Avoid diet rich in plant based fats
- Bile acid sequestrants: Cholestyramine
- Ezetimibe (selective cholesterol absorption inhibitor) - 10mg/day
- No use of statins as HMG CoA reductase activity is already inhibited
- Splenectomy if there is persistent anemia
Infantile Pyknocytosis
- Preterm infants presenting with transient hemolytic anemia (at 6-9 months of age) reticulocytosis and hyperbilirubinemia
- Peripheral smear- Pyknocytes- Distorted, densely stained RBCs
- Transfused RBCs also may become distorted
- No intervention needed
Rh Deficiency Syndrome
- Absence of Rh AG complex including Rh, RHAG, LW, Glycophorin beta, CD47 and protein 4.2
- Present with mild to moderate hemolytic anemia
- Peripheral smear- Stomatocytes, occasional spherocytes
- Treatment- Splenectomy- if hemolysis is severe.
Familial deficiency of HDL
- Accumulation of cholesterol in various tissues
- Large orange tonsils
- Hepatosplenomegaly
- Hemolytic anemia with stomatocytosis
Lecithin Cholesterol Acyl Transferase Deficiency
- Catalyses transfer of fatty acids from phosphotidylcholine to cholesterol
- Autosomal dominant
- Features include
- Hyperlipidemia
- Premature atherosclerosis
- Corneal opacities
- Chronic nephritis
- Proteinuria
- Mild anemia due to mild hemolysis
- Target cells are seen in peripheral smear
Abnormalities of Membrane Lipids
Acanthocytosis/ Spur cell anemia
- Greek: Acantha means thorn
- Spur cells are RBCs with irregular projections that vary in width, length and surface distribution.
- Cause: Severe liver disease due to any cause
- 2 step process:
- Outer lipid layer of the RBC membrane acquires additional nonesterified cholesterol from abnormal plasma lipoproteins
- Remodelling of abnormally shaped RBCs within spleen
- They undergo lysis in spleen as they are less deformable
- Present with progressive hemolytic anemia, splenomegaly, hepatic derangement
- Peripheral smear- RBCs show prominent, regular projection from surface.
- Splenectomy is potentially dangerous procedure in critically ill patients, hence is not recommended.
- Differential diagnosis- Zieves syndrome- Hyperlipoproteinemia, jaundice, spherocytic hemolytic anemia in alcoholic patients with liver disease
Abetalipoproteinemia
- Autosomal recessive codition
- Pathogenesis:
Defect in apolipoprotien B gene.
↓
Failure to synthesize/ secrete apolipoprotein
↓
No triglycerrides carried from intestines
↓
Decreased plasma triglyceride, cholesterol and phospholipid levels
↓
Increased sphingomyelin in outer membrane of RBCs
↓
Formation of acanthocytes/ burr cells
- It is often associated with
- Mild hemolysis
- Retinitis pigmentosa
- Progressive ataxic neurological disease with intentional tremors
- Fat malabsorption- Steatorrhea at 1 month of life
- Hepatic Encephalpathy
- Investigations:
- Peripheral smear: Acanthocytes are seen, Increased nucleated RBCs
- Reticulocytes are increased and they have normal shape
- Intestinal biopsy: Engorged mucosal cells with lipid droplets
- Treatment
- Dietary restriction of triglycerrides
- Supplement high doses of fat soluble vitamins (Vitamin A, D, E and K) and essential fatty acids.
Other neuro-acanthosis syndromes:
- Chorea acanthosis syndrome:
- Autosomal recessive
- Normolipoproteinemicacanthosis
- Neurological features include:
- Progressive orofacialdyskinesis with tics, limb chorea, lip and tongue biting
- Neurologic muscle hypotonia and atrophy
- Absent or diminished reflexes
- Increased CPK
- MRI- Abnormalities of putamen and head of caudate
- No anemia as RBC survival is mildly decreased
- Mechanism of acathocyte formation is not clearly known
- McLeod Phenotype
- Acanthosis together with decreased expression of Kell antigen (Kx protein)
- Defective gene on Xp 21(Close to genes of Duchene muscular dystrophy and retinitis pigmentosa)
- Gene codes for Kx protein that carries Kell blood group protein
- Males are affected
- Mild compensated hemolytic anemia
- May be associated with chronic granulomatous disease
- Often have myopathy/ chorea
- Patients may develop severe alloimmunization if transfused with Kell positive blood.
- Pantothenate kinase associated neurodegeneration (Hallervorden-Spatz syndrome)
- Progressive dementia, dystonia, spasticity, pallidial and retinal degeneration
- Allelic variant- HARP syndrome
- Hypobetalipoproteinemia
- Acanthosis
- Retinitis pigmentosa
- Pallidal degeneration
- Both are due to mutations in pantothenate kinase-2 gene
Figures:
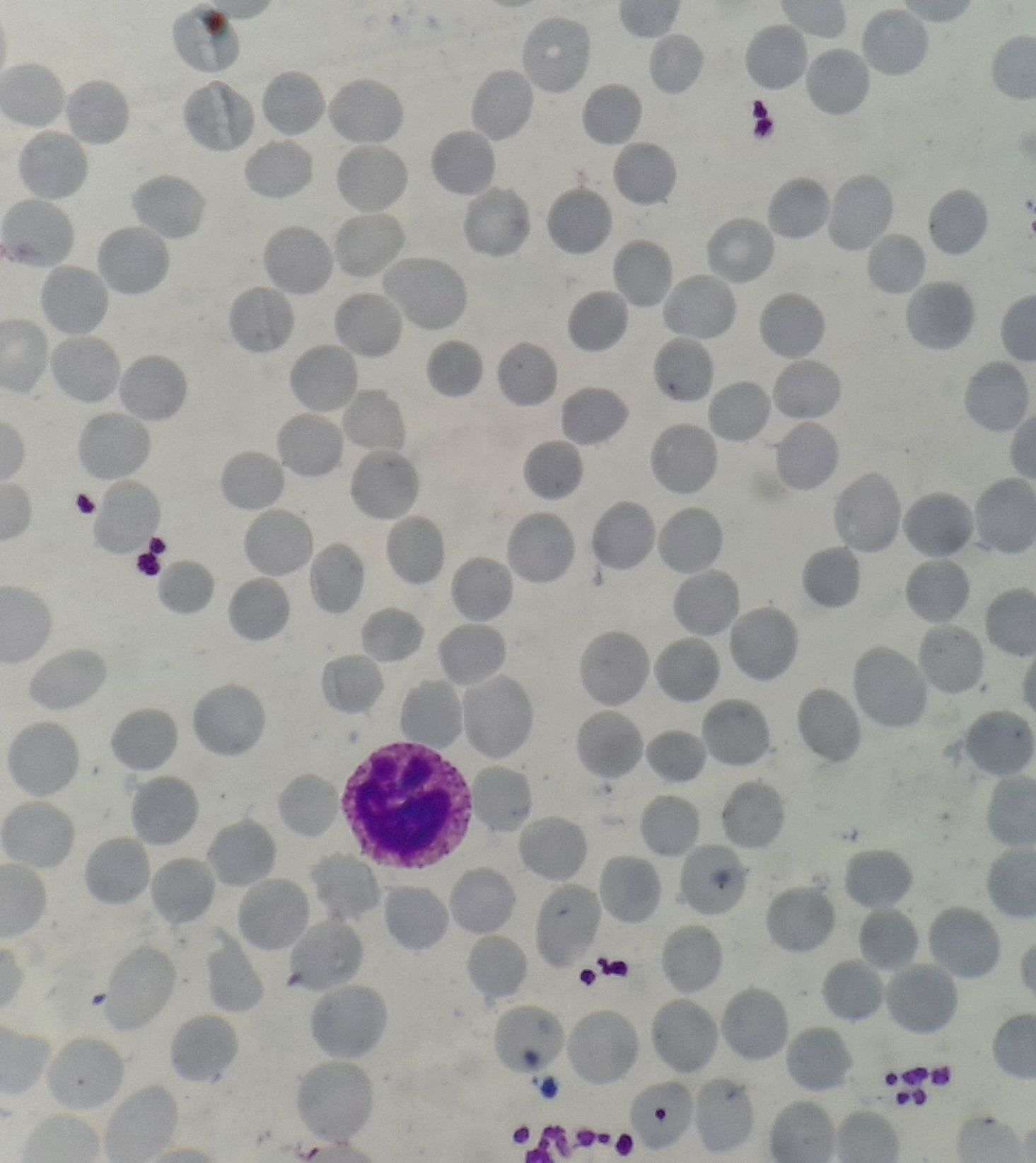
Figure 8.19.1- Hereditary spherocytosis- Peripheral smear
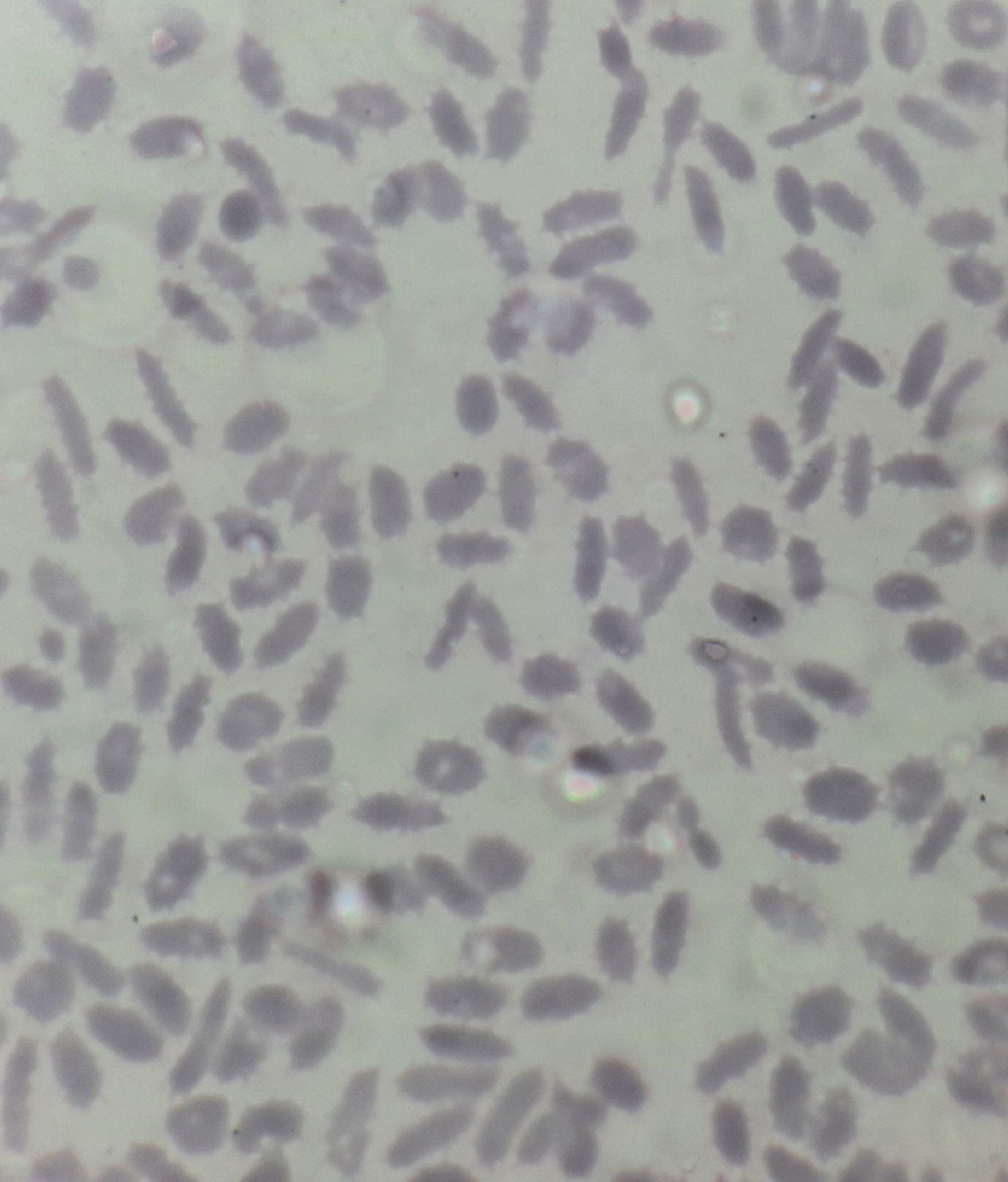
Figure 8.19.2- Hereditary elliptocytosis- Peripheral smear
An Initiative of
Veenadhare Edutech Private Limited
1299, 2nd Floor, Shanta Nivas,
Beside Hotel Swan Inn, Off J.M.Road, Shivajinagar
Pune - 411005
Maharashtra – India

howitreat.in
CIN: U85190PN2022PTC210569
Email: admin@howitreat.in
Disclaimer: Information provided on this website is only for medical education purposes and not intended as medical advice. Although authors have made every effort to provide up-to-date information, the recommendations should not be considered standard of care. Responsibility for patient care resides with the doctors on the basis of their professional license, experience, and knowledge of the individual patient. For full prescribing information, including indications, contraindications, warnings, precautions, and adverse effects, please refer to the approved product label. Neither the authors nor publisher shall be liable or responsible for any loss or adverse effects allegedly arising from any information or suggestion on this website. This website is written for use of healthcare professionals only; hence person other than healthcare workers is advised to refrain from reading the content of this website.