
howitreat.in
A user-friendly, frequently updated reference guide that aligns with international guidelines and protocols.
Myelodysplastic Neoplasms (MDS)
Introduction:
- It is a group of clonal hematological disorders characterized by moderate to marked dysplasia of one or more of 3 lineages (erythroid / myeloid / megakaryocytic), generally involving at least 10% of the cells of respective lineage in consideration, which results in cytopenias due to ineffective blood cell production.
- Defining cytopenia:
- Anemia: Hb <13 g/dL in males and <12 g/dL in females
- Leukopenia: ANC <1800/cmm
- Thrombocytopenia: Platelets <1,50,000/cmm
- Diagnostic evaluation must include:
- Peripheral smear
- Bone marrow
- Karyotyping,
- Mutation analysis by next-generation sequencing
- Multiparameter flow cytometry
Epidemiology:
- It is usually seen after age of 50 years.
- Median age of diagnosis is 70 years.
- Prevalence is 3-5 per 1 lakh in general population.
- In age group of more than 70 years prevalence is 20 per 1 lakh.
Etiology: In most of the cases cause is not known
- Exposure to radiation.
- Use of radiomimetic alkylating agents, topoisomerase inhibitors
- Smoking.
- Benzene, agricultural chemicals and solvents
- Congenital disorders like Down syndrome, Fanconi anaemia, Neurofibromatosis, Ataxia telangiectasia, xeroderma pigmentosa etc.
- Abnormalities in enzymes that metabolize carcinogens.
Pathogenesis:
- Increased apoptosis
- Mutations of proto-oncogenes such as MYC, TNER, tumour growth factor β &IL1 lead to increased apoptosis.
- BCL2 expression is also low in MDS.
- Increased apoptosis is seen in early phase of MDS especially in refractory anaemia & refractory anaemia with ringed sideroblasts.
- Increase in apoptosis is responsible for cytopenias in spite of hypercellular marrow.
- Apoptosis may be triggered by variety of factors such as.
- Intrinsic DNA damage.
- Mitochondrial dysfunction.
- Cytotoxic T cells.
- Over expression of cytokine TNF- with subsequent upregulation of the FAS/FAS L pathway.
- Abnormality of marrow stromal cells.
- Erythroid precursors in MDS spontaneously release cytochrome C from mitochondria, leading to activation of caspase 9.
- Immunological abnormalities in MDS
- These also explain presence of cytopenias in MDS
- Common features in aplastic anaemia and hypocellular MDS include.
- Clonal expansion of T-cells.
- Over representation of HLA-DR 15.
- Presence of mutations in PLG-A gene which is characteristic of PNH.
- Clinical response to immunosuppressive therapy.
- Decreased colony formation by hematopoietic stem cells
- Due to decreased production of GM-CSF, IL-3, M-CSF, and IL6.
- Epigenetic alterations:
- Methylation of DNA (especially of cytosine residues) leads to formation of CpG pair. Increased number of CpG islands proximal to gene promoter region causes decreased gene expression (gene silencing).
- Silencing of tumour suppressor genes such as CTNNA1, P15INK4B lead to increased proliferation of cells.
- Formation of ringed sideroblasts.
Mitochondrial DNA mutations are seen in 50% of MDS cases.
Impaired function of mitochondrial respiratory chain
↓
Oxidation of ferrous ion to ferric form
↓
Accumulation of ferric ions in mitochondria
↓
Formation of ringed sideroblasts
- Change from MDS to AML occurs due to accumulation of additional mutations which block the maturation at blast level.
New (2024) WHO Classification: (Details given at the end)
Disease | Blasts | Cytogenetics | Mutations |
MDS with defining genetic abnormality | |||
MDS with low blasts and 5q deletion | <5% BM and <2% PB | 5q deletion alone, or with 1 other abnormality other than monosomy 7 or 7q deletion | Any except multi-hit TP53 |
MDS with low blasts and SF3B1 mutation (MDS with low blasts and ringed sideroblasts) | <5% BM and <2% PB | Absence of 5q deletion, monosomy 7, or complex karyotype | SF3B1 (Detection of ≥15% ring sideroblasts may substitute for SF3B1 mutation) |
MDS with biallelic TP53 inactivation | <20% BM and PB | Usually complex | Multihit TP53 alterations (Two or more TP53 mutations, or 1 mutation with evidence of TP53 copy number loss or copy neutral Loss of Heterozygosity) |
MDS, morphologically defined | |||
MDS with low blasts (MDS-LB) | <5% BM and <2% PB | Any | Any except multihit TP53 or SF3B1 |
MDS, hypoplastic (MDS-h) | <5% BM and <2% PB | Any | Any except multihit TP53 or SF3B1 |
MDS with increased blasts- 1 | 5-9% BM or 2-4% PB | Any | Any except multihit TP53 or SF3B1 |
MDS with increased blasts- 2 | 10-19% BM or 5-19% PB or Auer rods | Any | Any except multihit TP53 or SF3B1 |
MDS with fibrosis | 5-19% BM; 2-19% PB | Any | Any except multihit TP53 or SF3B1 |
Old Classification:
Disease | Blood Findings | Bone Marrow Findings |
Refractory cytopenia with unilineage dysplasia | Uni/Bicytopenia No or rare blasts | <5% blasts <15% ringed sideroblasts Dysplasia in only one lineage of cells. 3 subtypes: 1. Refractory anemia- Dysplasia in erythroids 2. Refractory neutropenia- Dysplasia in myeloid cells 3. Refractory thrombocytopenia- Dysplasia in megakaryocytes (>10% dysplastic megakaryocytes of at least 30 megakaryocytes evaluated) |
Refractory anemia with ringed sideroblasts (RARS) | Anemia No blasts | >15% ringed sideroblasts Erythroid dysplasia only <5% blasts |
Refractory cytopenia with multilineage dysplasia (RCMD) | Cytopenias (bicytopenia or pancytopenia) No or rare blasts No Auer rods < 1 x 109/L monocytes | Dysplasia in >10% of the cells of two or more myeloid cells lines <5% blasts in marrow No Auer rods |
MDS with excess blasts -1 | Cytopenias <5% blasts No Auer rods < 1 x 109/L monocytes | Unilineage or multilineage dysplasia 5-9% blasts No Auer rods |
MDS with excess blasts -2 | Cytopenias 5-19% blasts Auer rods +/- < 1 x 109/L monocytes
Presence of Auer rods in blasts in PB/BM qualifies a case as RAEB-2 irrespective of blast percentage. | Unilineage or multilineage dysplasia 10-19% blasts Auer rods +/- |
Myelodysplastic syndrome unclassified (MDS-U) | Cytopenias No or rare blasts No Auer rods | Unilineagedysplasia in<10% of cells in one or more myeloid cell lines <5% blasts No Auer Rods |
MDS associated with isolated del(5q) | Macrocytic anemia Usually normal or increased platelet count <1% blasts | Normal to increased megakaryocytes, which are slightly smaller in size. Nucleus is hypolobated and eccentrically placed. Dysmorphic erythropoiesis is seen in some cases (Lobulated erythroblast nuclei) <5% blasts Isolated del(5q) cytogenetic abnormality No Auer rods |
If SF3B1 mutation is present, then presence of >5% sideroblasts are enough to classify it as MDS with ringed sideroblasts
Note: Presence of MDS related mutations alone (with absence of morphologic dysplasia), even in the presence of clinically significant cytopenias, is not diagnostic of MDS.
Clinical Features:
- Asymptomatic
- Anaemia
- Recurrent infections due to leukopenia. Bacterial pneumonia and skin abscess are most common.
- Haemorrhagic manifestation – Occurs due to thrombocytopenia or abnormal platelet function.
- Skin lesions – Sweet’s syndrome (Febrile neutrophilic dermatosis), pyoderma gangrenosum, cutaneous vasculitis.
- Wide range of auto immune disorders seen in 10% cases- Cutaneous vasculitis, relapsing polychondritis, polymyalgia rheumatica, necrotizing panniculitis, Coomb’s positive auto immune haemolytic anaemia, seronegative synovitis and arthritis etc
- Hepatomegaly in 5% cases
- Splenomegaly in 10% cases
Investigations:
- Hemogram:
- Normocytic / macrocytic hypochromic anaemia
- Anisopoikilocytosis, oval macrocytes
- Howell – jolly bodies may be seen which indicate accelerated erythropoiesis.
- Basophilic stripling.
- Nucleated RBCS seen.
- Sideroblasts may be seen.
- Reticulocytopenia may be noted.
- Neutropenia may be noted
- Neutrophils are hypogranular, hyposegmented; contain Dohle bodies, sometimes Auer rods.
- Pseudo Pelger Huet anomaly– They are neutrophils with bilobed nuclear configuration
- Nuclear fragmentation/ ring shaped nuclei / nuclear sticks may be seen
- Monocytosis may be seen in some cases
- Rarely circulating myeloblasts may be seen
- Thrombocytopenia / thrombocytosis
- Platelets are large and lack granules
- Circulating micromegakaryocytes may be present. They characteristically contain cytoplasmic blebs and sometimes 1-2 platelets are attached to the surface.
- Percentage of blasts must include all nucleated cells except nRBCs.
- Reticulocyte count- Inappropriately low
- Serum iron, ferritin level – Increased
- Serum cobalamin and folic acid levels – Increased
- Bone marrow aspiration
- Cellularity – Normal / increased / decreased (If hypocellular, dysplasia must be present in myeloid precursors and megakaryocytes for diagnosis of MDS, as erythroid dysplasia is often present in aplastic anaemia)
- Dyserythropoiesis:
- Megaloblastoid changes
- Nucleus – Nuclear fragmentation, nuclear budding, abnormal nuclear shape, karyorrhexis, irregular staining properties, internuclear bridging, multinuclearity, and nuclear hyperlobation.
- Cytoplasm – vacuolation, defective haemoglobinisation, and basophilic stripling are noted.
- Giant, multinucleated erythroid precursors may be noted
- Ringed sideroblasts are present – They are the normoblasts in which mitochondrial iron deposits. By definition, they have 5 or more iron granules, that encircle one third or more of the nucleus in an iron stained (Prussian blue reaction) smear.
- Dysgranulopoiesis:(Abnormal granulocyte maturation)
- Abnormal staining of primary granules in promyelocyte and myelocyte. Sometimes these granules are larger (Pseudo- Chediak- Higashi granules).
- Secondary granules may be absent in myelocyte and mature neutrophils.
- Irregular cytoplasmic basophilia with a dense rim of peripheral basophilia is also characteristic.
- Auer rods are sometimes seen.
- Nuclear hypolobulation or hypersegmentation may be noted.
- Increased number of marrow monocytes.
- In case of del (17p)- Hypolobated neutrophil nuclei are seen
- Blast Cells- Maximum number of blast cells compatible with a diagnosis of MDS is 20%. Blast count is an important prognostic indicator in MDS
- Blasts have central nucleus with fine nuclear chromatin, prominent nucleoli, high nuclear – cytoplasmic ratio, deeply basophilic and a granular cytoplasm. Auer rods may be seen in some blasts.
- Percentage of blasts must be percentage of all nucleated cells cells in BM (including erythroid precursors)
- Dysmegakaryopoiesis
- Micromegakaryocytes with nuclear hypolobation
- Megakaryocytes with small, multiple, separated nuclei
- Large mononuclear megakaryocytes
- Granules may be absent or may be large and abnormal.
- Trephine Biopsy
- It is done to detect "Abnormal localization of immature precursors" (ALIP).
- Presence of ALIP indicates increased risk of transformation to leukaemia
- Normally immature myeloid precursors are seen close to bony trabeculae and around blood vessels, whereas developing erythroid cells and megakaryocytes are seen in inter trabecular spaces
- In ALIP, myeloid precursors are displaced from trabecular margins and small clusters of myeloblasts and promyelocytes are seen in inter trabecular spaces
- 5 cell clusters and 3 clusters in single HPF are significant.
- Hypocellularity can be better appreciated in BM biopsy sample. <10% cellularity indicates hypocellular MDS. Always rule out toxic myelopathy and autoimmune disorders in such patients.
- Cytochemistry
- Peroxidase and Sudan black B- Positive in blasts of myeloid origin
- Iron stains- Helps in identification of ringed sideroblasts
- PAS- Positive in erythroid precursors
- Flow cytometry abnormalities
- Low side scatter due to cytoplasmic hypogranularity. Differentiated from myeloblasts by lack of CD34, CD117 and/or HLA-DR. MDS cells are often positive for CD10 and CD16.
- Down regulation of antigens that are normally expressed on myeloid cells
- Abnormal patterns of cell marker expression
- Lineage infidelity in antigen expression.
- Increased ratio of CD34+/CD33+
- Premature phenotype of myeloid cells- Increased expression of CD33 and CD13 with loss of CD16.
- NAT 9- Decreased expression
- Large granular lymphocyte population may be identified with flow.
- Evaluation of PNH clone and HLA-DR15can be done with flow. If they are positive, patients are responsive to immunosuppressive therapy.
- Flow helps to know antibody combinations that characterize the blasts.
- Flow percentage of CD34+ cells cannot replace differential count on smears while calculating percentage of blasts.
- Cytogenetics
- Abnormalities are found in 50% of cases.
- Essential investigation in MDS, as it helps to prove clonality and important in prognostication.
- Should be performed on at least 25 metaphases.
- If there is a dry tap, do FISH on peripheral blood for monosomy 7, del5q and trisomy 8.
- Common abnormalities include:
- Deletion of 5q/ Monosomy 5
- Deletion of 7q / Monosomy 7
- Trisomy 8
- t (11q23)
- del 17p
- del 20q
- del 18q
- Complex karyotyping abnormalities
- Molecular studies by next generation sequencing: Mutation of following genes may be seen
- N-RAS – Oncogene
- P53, IRF1 – Tumor suppressor genes
- BCL-2- Antiapoptotic gene
- CSF – 1R & AML -1 gene
- TET2, AXSL1, RUNX1, TP53, EZH-2, NRAS, JAK2, ETV6, CBL, IDH2
- Duplication of FLT-3 gene
- Methylation of CpG islands in promoter regions of genes
- Hypermethylation of p15 gene – p15 encodes an inhibitor of cell cycle progression.
- Shortened telomeres, increased telomerase activity and microsatellite instability
- Response of T-lymphocytes to mitogens PHA and Con A- Reduced
- Granulocytic oxidative metabolism test. (Measured by nitroblue tetrazolium reduction test) – Abnormal
- Platelet function test – Reduced aggregation with adrenaline and collagen
- Platelet associated immunoglobulin – May be raised in the absence of immune mediated thrombocytopenia
- Immunoglobulin study
- Polyclonal gammopathy in 30%
- Hypogammaglobulinemia in 20%
- Paraproteins in 5-10%
- Copper level- Deficiency may mimic MDS
- LDH and Uric acid levels- Increased
- Beta 2 microglobulin level- May be increased
- MRI of femoral head- Hyperintensity
- S. Erythropoietin levels- Helpful in planning the treatment in refractory anaemia
- TSH- To rule out hypothyroidism as cause of anemia
- HLA typing- If transplant is planned
- Genetic screening for Fanconi anaemia, dyskeratosis congenita, RUNX1 and GATA2 in young patients
- HbF level-Elevated in 5% cases
- Acquired HbH disease in some cases leading to microcytic hypochromic anaemia
Criteria for Diagnosis:
- To establish diagnosis of MDS, careful morphologic review and correlation with patient's clinical features are important, as number of medications and viral infections may cause morphologic changes in marrow cells similar to MDS.
- Diagnosis is established when
- Other causes of dysplasia are excluded
- Cytopenia persists for at least 6 months
- To call as significant dysplasia is present, ≥10% cells of respective lineage must show features of dysplasia or Blast count is 5-19% or an MDS associated karyotype must be present
MDS-defining abnormalities (by conventional cytogenetics): −7 or del(7q), t(11;16)(q23;p13.3), −5 or del(5q), t(3;21)(q26.2;q22.1), i(17q) or t(17p), t(1;3)(p36.3;q21.1), −13 or del(13q), t(2;11)(p21;q23), del(11q), inv(3)(q21q26.2), del(12p) or t(12p), t(6;9)(p23;q34), del(9q), idic(X)(q13), or complex karyotype (three or more chromosomal abnormalities involving one or more of the above).
Prognosis:
International Prognostic scoring system (IPSS)
Score value | 0 | 0.5 | 1.0 | 1.5 | 2.0 |
BM blasts (%) | <5 | 5-10 | - | 11-20 | 21-30 |
Karyotypes* | Good | Intermediate | Poor |
|
|
Cytopenias | 0/1 | 2/3 |
|
|
|
*Good: Normal, -Y alone, del (5q) alone, del (20q) alone
Poor: complex (≥3 abnormalities) or chromosome 7 anomalies
Intermediate: other abnormalities.
Cytopenia defined as hemoglobin concentration <10 g/dL, neutrophils<1.5 x 109/L and platelets < 100 x 109/L
Median survival of primary myelodysplastic syndrome using the IPSS score.
Risk Group | IPSS score | Median Survival (in years) | |||
<60 years | > 60 years | <70 years | > 70 years | ||
Low | 0 | 11.8 | 4.8 | 9 | 3.9 |
Intermediate 1 | 0.5-1.0 | 5.2 | 2.7 | 4.4 | 2.4 |
Intermediate 2 | 1.5-2.0 | 1.8 | 1.1 | 1.3 | 1.2 |
High | >2.5 | 0.3 | 0.5 | 0.4 | 0.4 |
- Revised IPSS scoring considers anaemia, neutropenia and thrombocytopenia as separate entities. Also, classification of cytogenetic abnormalities is different.
- Other scoring systems include Düsseldorf, Varela, Sanz and Bournemouth
Adverse prognostic factors in MDS
- Severe cytopenias
- Raised LDH or beta 2– microglobulin
- Increased blasts
- Trilineage dysplasia
- Presence of ALIPs
- Chromosome abnormalities
- Loss of chromosome 5 or 7
- Deletion of chromosome 3q, 5q (excluding 5q syndrome), 7q, 17p
- Structural abnormality of chromosome 11q23
- Complex chromosome abnormalities
- P53, RAS mutations
- Over-expression of WT1
- P15 hypermethylation
- Mutations of ASXL1, RUNX1, TP53, EZH-2, NRAS, ETV6 (High risk of leukemic transformation)
- Immunophenotype- CD7-Positive blasts
- Telomere shortening
In general
- Median survival for all MDS is less than 2 years
- Leukemic transformation occurs in 10-40% cases
Differential Diagnosis: Other causes for marrow dysplasia
(Slides for assessing marrow dysplasia should be of very high quality. They should be prepared from freshly obtained specimen. Presence of clonality, rules out all below mentioned possibilities and establishes diagnosis of MDS. Hence doing cytogenetics is must in all suspected cases of MDS)
- Healthy elderly individuals.
- Vitamin B-12 and folic acid deficiency
- Heavy metal- Especially arsenic and zinc
- Alcohol abuse
- HIV and Parvo virus infection
- Antituberculous therapy
- Treatment with G-CSF
- Congenital dyserythropoieticanaemia
- Drugs and chemicals- Cotrimoxazole, mycophenolate, chemotherapy agents, valproic acid
- PNH
- Copper deficiency
- Chronic liver disease
Pretreatment Work-up:
- History- Transfusions
- Examination
- WHO P. S.
- BSA
- FCM (for MDS, LGL and PNH)
- BMA and Bx
- BMA Iron stain
- Haemoglobin
- TLC, DLC
- Platelet count
- Peripheral smear
- Reticulocyte count
- S. EPO levels (for RA)
- Iron study: Ferritin: S. Iron: TIBC: T.Sat:
- S. B12 level
- RBC Folic acid level
- R/O Copper Deficiency- Malabsorption/ Gastric bypass/ Zn supplementation
- LFT- Bili- T/D SGPT: SGOT:
- Creatinine
- Uric acid
- LDH
- HIV
- HBsAg
- HCV
- UPT
- Cytogenetics
- FISH MDS Panel
- Molecular testing for MDS related genes (selected cases)
- RUNX1 OR GATA 2 in familial cases
- Prognostic score
- HLA typing for HSCT candidate
- HLA-DR 15 typing (for hypo MDS)
- Chemotherapy consent after informing about disease, prognosis, cost of therapy, side effects, hygiene, food and contraception
- Fertility preservation
- PICC line insertion and Chest X ray after line insertion
- Tumor board meeting and decision
- Attach supportive care drug sheet
- Inform primary care physician
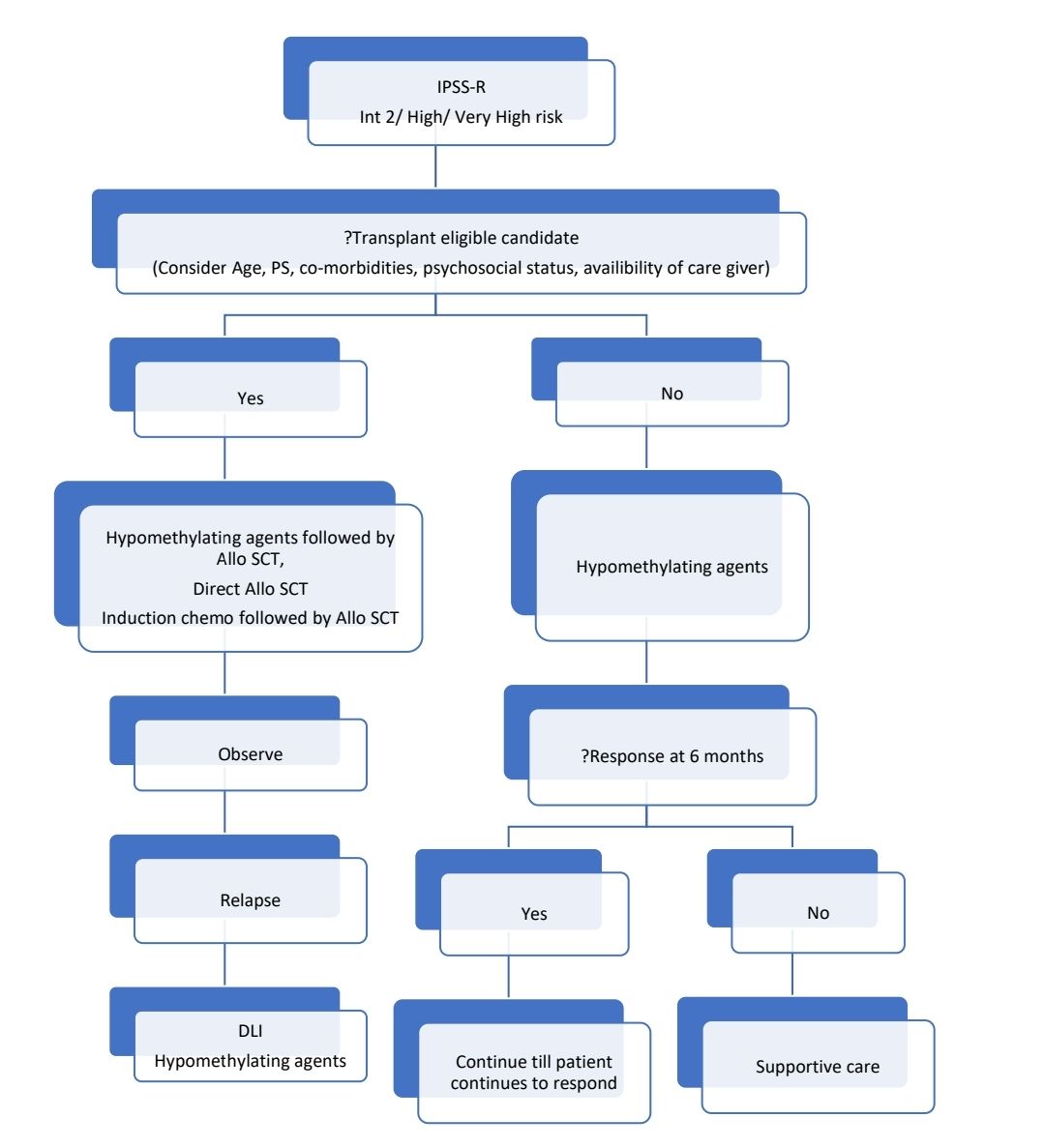
Treatment Plan:
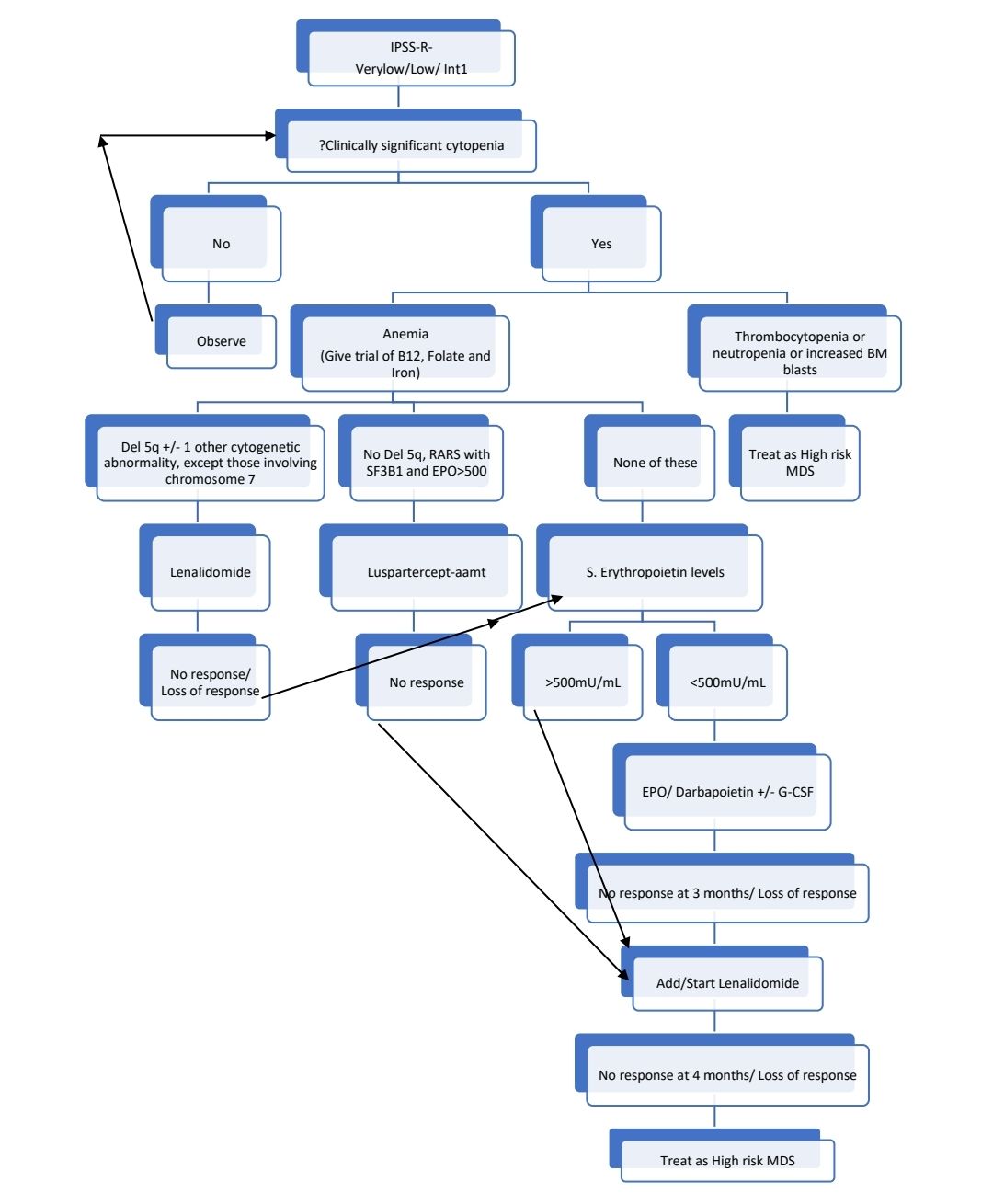
For following category of patients consider ATG with Cyclosporine therapy:
- <60 years with hypocellular BM with <5% blasts
- PNH clone positivity
- STAT-3 mutant cytotoxic T cell clones
International Working Group Modified Response Criteria:
- CR Marrow- ≤5% blasts in BM and normal maturation of all cell lines
- CR Peripheral blood
- Hemoglobin ≥ 11gm/dL
- ANC ≥ 1000/cmm
- Platelets ≥ 1lac/cmm
- No blasts in peripheral smear
Criteria for hematological improvement:
- Rise in hemoglobin by 1.5gm/dL
- Platelet count >30,000/cmm
- ANC >500/cmm
About Each Modality of Treatment:
- Hypomethylating agents:
- Disease modifying agents which offer survival advantage but not cure
- Induce demethylation of hypermethylated genes.
- Usually 4-6 cycles are given, then if there is response, then hypomethylating agents are continued till there is loss of response
- 30% patients show haematological response.
- Results in improved quality of life.
- They also delay transformation to AML.
- No survival advantage over supportive care
- Azacytidine is preferred over Decitabine
- Patients failing response to one type of hypomethylating agent, may respond to other.
- High dose vitamin C can improve response to Azacytidine.
- Hypomethylating agents can be used as interim therapy till transplant arrangements are made. They can used to induce remission as well, in patients who are not fit for 7+3 induction
- Decitabine
- Inj. Decitabine- 20mg/m2- in 300ml NS over 3 hrs- From Day 1 to Day 5
- Frequency: 28 days
- Dose adjustments:
- S. Creatinine- >2mg/dL- Hold until recovery of toxicity
- Bilirubin- >2gm/dL- Hold until recovery
- ANC <1000/cmm or platelet count <50,000/cmm- Hold until CBC values increase to more than cut off values (this applies only if, baseline ANC was >1500/cmm and baseline platelet count was >75,000/cmm)
- Active infection- Hold until recovery
- Azacytidine
- Inj. Azacytidine- 75mg/m2- SC- OD- Divide doses if volume is >4ml.- From Day 1 to Day 7
- Frequency: 28 days
- Dose adjustments:
- S. Creatinine- >3mg/dL- Hold until recovery to normal and then restart with 50% of dose.
- Hepatic impairment- Use with caution
- ANC <1000/cmm or platelet count <50,000/cmm- Hold untill CBC values increase to more than cut off values and then give 50% of dose. (this applies only if, baseline ANC was >1500/cmm and baseline platelet count was >75,000/cmm)
- Active infection- Hold until recovery
- Much lower doses have also been tried which has shown less toxicity and more effectiveness
- Decitabine- 20mg/m2 OD for 3 days in 46 days cycle
- Azacytidine- 20-45mg/m2-OD 3-5 days, every 4-6 weeks
- Lenalidomide:
- Dose- 10mg- OD for 21 days in 28 days cycle. (Dose of 5mg- OD is tolerated by many patients)
- Response is assessed at 2-4 months
- No response is <1.5gm/dL rise in haemoglobin or transfusion dependency at 12-16 weeks of therapy.
- Luspatercept: Details are available in drugs section. Click here
- Erythropoiesis stimulating agents (ESA)
- Erythropoietin- 40,000 units- SC- Once a week, if no response at 8 weeks, increase dose to 60,000 units per week for further period of 8 weeks.
- Darbepoetin- 300mcg SC once in 2 weeks, if no response at 8 weeks, increase the dose to 300mcg- SC- Once a week for further period of 8 weeks.
- If there is no response (complete/partial) after 16 weeks of therapy then stop erythropoietin. Complete response is achievement of hemoglobin >11gm/dL and transfusion independence. Partial response is rise in hemoglobin by >2gm/dL from baseline (but less than 11gm/dL) and transfusion independence.
- Rule out hemolysis and deficiency status prior to starting erythropoietin
- Good response in following situation
- Low serum EPO levels - < 500 U/L
- Non RARS subtype
- No / Low transfusion requirements
- RARS Subtype respond well when EPO is combined with G-CSF
- In those with durable complete response the dose of ESA should be slowly reduced, to a lowest dose that maintains the response.
- If response is lost, consider functional iron deficiency.
- Avoid rise of haemoglobin to >12gm/dL, as there is high risk of thrombosis (especially if there is previous stroke or history of DM2/ Hypertension)
- G-CSF
- 90% show – Dose dependent neutrophilia and improvement in neutrophil function
- Given to patients with severe sepsis
- Adjust the dose to maintain TLC between 6000 - 1000/cmm.
- Start with 300mcg SC once a week, if there is no response increase the dose up to 300mcg SC 3 times a week.
- It does not affect the risk of leukemic transformation
- Eltrombopag
- It is useful in following conditions
- Positive for PNH clone
- Age <60 years
- Hypoplastic MDS
- HLA-DR 15 positive patients
- It is useful in following conditions
- Intensive chemotherapy
- It is given to reduce the tumour burden, so that risk of relapse is less after transplant.
- Useful if stem cell transplant is planned following cytoreduction.
- Donor must be identified prior to starting induction chemotherapy, if time allows.
- Usually 7+3 induction chemotherapy is given
- Remissions are seen in less than 20% of patients
- It should be avoided in patients with poor performance score, as this therapy worsens patient’s condition.
- It should be considered in patients less than 50 years of age.
- Factors association with higher chances of CR
- Early treatment (within 3 months of diagnosis)
- Young age (<50 years)
- Normal Karyotype
- Presence of Auer rods
- Allogeneic Stem cell transplantation
- It is the only curative treatment
- Conditioning is usually done with Busulfan and Cyclophosphamide.
- PBSC is better than BM harvest
- Overall disease-free survival- 30-40%
- Given after induction of CR by hypomethylating agents or induction chemotherapy. If marrow blast count is <5% then, prior chemotherapy is not required.
- Good results with
- Patients < 40 years old
- Short disease duration
- <5% BM blasts
- Platelet count >1lac/cmm
- Poor prognostic factors for outcome
- Old age
- Poor risk cytogenetics
- High IPSS score
- Advanced disease (RAEB, RAEBt)
- Treatment related MDS
- Prolonged disease duration
- Marrow fibrosis
- Reduced intensity conditioning:
- Least day 100 mortality
- 2-year event free survival- 56%
- With RIC, it is possible to transplant patients aged >70 years
- Disease control depends on graft vs. MDS effect
- For relapse after Allo SCT following can be done:
- Salvage with DLI
- Second transplant
- Chemotherapy
- Results with MSD and MUD are same. Hence if familial bone marrow failure syndromes are suspected, MUD is preferred over MSD.
- Autologous transplant has problem of contamination of stem cell product with tumour cells. There is no GvL effect as well. Hence there are high chances of relapse. There is 10% peritransplant mortality. With advent of RIC, it is not recommended now.
- Immunosuppression with ATG and Cyclosporine
- Better response in case of – Hypocellular MDS and HLA – DR15 positive cases
- Response rate in unselected MDS- 30 – 50%
- ATG must be restricted to fit, relatively young patients.
- Dose and method of administration is similar to that in aplastic anaemia.
- Ciclosporine is continued for at least 6 months aiming a trough level of 100-200 microgm/L and then slowly tapered
Supportive Care:
- Red cell transfusion of correct anaemia. Trigger depends on patient’s activity level. Generally, it is better to maintain haemoglobin level of 7-8gm/dL.
- Chelation is given once patient has received 5g iron (25 units of red cells) & those who have a good prognosis according to IPSS score and reasonable life expectancy.
- Platelet transfusions for thrombocytopenia
- Antibiotics to prevent infections in case of neutropenia
Other Treatment Options:
- Low dose cytarabine
- May be useful in intermediate 2 and high risk patients
- 5-20mg/m2/day- SC-BD- for up to 8-16 weeks
- Remission is seen in about 20% patients, which remains for about 10 months
- Survival is same as supportive care alone
- Danazol- Useful in increasing platelet count in some cases
- Hydroxyurea and Low dose etoposide- Control leukemic cell population but does not influence survival duration
- Maturation enhancing agents
- Retinoids
- Vitamin D derivatives
- Arsenic trioxide
- Azacytidine with Lenalidomide
- Histone deacetylase inhibitors- Vorinostat and others
- Luspatercept, Sotatercept, Roxadustat
- Low dose Melphalan- 2mg/day for 50 days. CR observed in 30% patients which remains for approximately 1 year.
Subtypes of Myelodysplastic neoplasms:
MDS with defining genetic abnormalities
MDS with low basts and 5q deletion
- It is a myeloid neoplasm with cytopenia and dysplasia characterized by chromosome 5q deletion occurring in isolation or with one other cytogenetic abnormality (other than monosomy 7 or 7q deletion).
- Account for 2.5% of all MDS
- Median age- 67 years
- Common in women
- Present with anemia requiring regular transfusions
- Peripheral smear:
- Macrocytic anemia
- Usually normal or increased platelet count
- <1% blasts
- Bone marrow
- Normocellular or hypercellular
- Erythropoiesis is suppressed
- Dysmorphic erythropoiesis is seen in some cases (Lobulated erythroblast nuclei)
- Granulocytic dysplasia is uncommon
- Normal to increased megakaryocytes, which are slightly smaller in size. Nucleus is hypolobated and eccentrically placed.
- <5% blasts
- No Auer rods
- Presence of ringed sideroblasts/ SF3B1 mutation does not exclude the diagnosis of MDS with 5q deletion
- Prognosis:
- Most patients belong to IPSS- low risk
- TP53 mutation is associated with decreased response to lenalidomide and increased risk of transformation to AML
MDS with low blasts and SF3B1 mutation
(MDS with low blasts and ringed sideroblasts)
- It is a myeloid neoplasm with cytopenia and dysplasia characterized by SF3B1 mutation and ring sideroblasts.
- Account for 17% of all MDS cases
- Median age- 70- 75 years
- Present with chronic anemia
- Peripheral smear
- Macrocytic or normocytic normochromic RBCs
- Rarely neutropenia and thrombocytopenia
- Blasts are rarely seen
- Granulocytes may show dysplastic features
- Bone marrow
- Hypercellular
- Increased erythropoiesis with dysplastic features
- Ringed sideroblasts are seen in iron stain
- Dysplastic changes in myeloid cells may be present
- Megakaryocytes are normal
- Blasts - <5%
- Cytogenetics: Usually normal
- Criteria for diagnosis: Essential
- Cytopenia involving one or more lineages, without thrombocytosis
- Erythroid lineage dysplasia;
- Blasts <5% in the bone marrow and <2% in the peripheral blood;
- Detection of SF3B1 mutation (High- >5% VAF is necessary). If SF3B1 mutation analysis is not available, demonstration of ring sideroblasts comprising ≥15% of erythroid precursors
- Not fulfilling diagnostic criteria of AML, MDS with low blasts and 5q deletion, MDS with biallelic TP53 inactivation, MDS with increased blasts, or any MDS/MPN type.
- Prognosis: Best among all MDS subtypes. Most belong to IPSS-Low risk.
MDS with biallelic TP53 inactivation
- It is a myeloid neoplasm with cytopenia, dysplasia and less than 20% blasts or 30% erythroblasts, characterized by two or more TP53 mutations or one TP53 mutation and concurrent evidence of TP53 copy loss or copy neutral loss of heterozygosity
- Account for 7-11% of all MDS
- Present with pancytopenia
- Bone marrow
- Dysplasia noted in all 3 lineages
- Blast count is usually high
- BM fibrosis if often present
- Biallelic alterations can be identified by NGS. TP53 VAF of >40% is considered as significant.
- Cytogenetics: Usually complex abnormalities are found
- Criteria for diagnosis:
- Essential:
- Cytopenia involving one or more lineages
- Dysplasia involving one or more lineages
- Blasts constitute <20% in PB and BM
- Detection of one or more TP53 mutations
- In the presence of one TP53 mutation, evidence of TP53 copy loss or copy neutral LOH
- Desirable:
- Complex karyotype (at least 3 abnormalities)
- Essential:
- Prognosis: Risk of leukemic transformation and hence death is high.
MDS, morphologically defined
MDS with low blasts (MDS-LB)
(MDS with single lineage dysplasia and MDS with multilineage dysplasia)
- It is a myeloid neoplasm with cytopenia and dysplasia but without defining genetic abnormalities, defined by having <5% bone marrow blasts and <2% peripheral blood blasts.
- Accounts for 15- 20% of all MDS cases
- Most of them present with anemia
- PS and BM findings: As described above
- Cytogenetics: Usually normal
- Criteria for diagnosis:
- Essential:
- Cytopenia involving one or more lineages.
- Dysplastic changes in one or more lineages, involving at least 10% of cells.
- <5% bone marrow blasts and <2% peripheral blood blasts
- Exclusion of folate and vitamin B12 deficiency.
- No fulfilling diagnostic criteria of MDS with defining genetic alterations or hypoplastic MDS.
- Desirable:
- Hypercellular bone marrow for age.
- Detection of clonal cytogenetic and/or molecular abnormality.
- Essential:
- Prognosis: Depends on type and degree of cytopenia
MDS, hypoplastic (MDS-h)
- It is a myeloid neoplasm with cytopenia and dysplasia, characterized by significantly decreased age-adjusted bone marrow cellularity as determined on a trephine biopsy.
- Accounts for 10-15% of all MDS.
- Significant overlap with PNH and aplastic anemia is noted.
- They have more profound cytopenia, compared to other MDS.
- Bone marrow:
- Cellularity: <30% in patients younger than 70 years and below 20% in patients aged 70 and older.
- Dysplastic features are identified in one or more haematopoietic lineages.
- Cytogenetics: Abnormal in 25-40% cases. Commonly trisomy 8 and del (20q).
- Criteria for diagnosis:
- Essential:
- Cytopenia involving one or more lineages
- Hypocellular bone marrow (assessed on a trephine core biopsy, adjusted for age of the patient) not explained by drug/toxin exposure or pertinent nutritional deficiency or PNH or inherited BM failure syndromes
- Dysplasia involving myeloid and/or megakaryocytic lineages
- <5% blasts in bone marrow and <2% blasts in peripheral blood
- Not meeting criteria for MDS with defining genetic abnormalities or MDS with increased blasts.
- Desirable:
- Detection of clonal cytogenetic and/or molecular abnormality.
- Essential:
- Prognosis: Significantly worse compared to aplastic anemia, but better, when compared with other MDS.
- Treated usually with immunosuppressive therapy
MDS with increased blasts
- It is a myeloid neoplasm with cytopenia and dysplasia but without defining genetic abnormalities, defined by having 5-19% bone marrow blasts and/or 2-19% peripheral blood blasts.
- 2 subtypes:
- MDS with increased blasts- 1: Blasts: 5-9% BM or 2-4% PB
- MDS with increased blasts- 2: Blasts: 10-19% BM or 5-19% PB or Auer rods
- MDS with increased blasts and fibrosis (MDS-F): 5-19% blasts in the bone marrow and/or 2-19% blasts in the peripheral blood, with significant bone marrow fibrosis (defined as grade 2 or 3).
- Accounts for 29-38% of all MDS cases
- Most present with pancytopenia
- Cytogenetics: Abnormal in 50- 70% cases
- Criteria for diagnosis:
- Essential:
- Cytopenia involving one or more lineages
- Dysplastic changes in one or more lineages, involving at least 10% of cells
- 5-19% bone marrow blasts and 2-20% peripheral blood blasts
- Not fulfilling diagnostic criteria of MDS with biallelic TP53 inactivation or AML.
- Desirable:
- Detection of clonal cytogenetic and/or molecular abnormality.
- Essential:
Childhood Myelodysplastic Neoplasm
(Refractory cytopenia of childhood)
- It is a myeloid neoplasm with cytopenia and dysplasia arising in children and adolescents (<18 years of age)
- 3 subtypes:
- Childhood myelodysplastic neoplasm with low blasts: <5% bone marrow blasts and <2% peripheral blood blasts.
- Childhood myelodysplastic neoplasm with increased blasts: 5-19% bone marrow blasts and/or 2-19%% peripheral blood blasts
- Childhood myelodysplastic neoplasm with low blasts- hypocellular
- Incidence-
- 1-4 cases/Million population/ year
- Median age- 6.8 years
- Strongly associated with congenital disorders such as
- Congenital aplastic anaemia syndromes
- Neurofibromatosis- type I
- Bloom syndrome
- Noonan syndrome
- Dubowitz syndrome
- Cytogenetic changes
- Monosomy 7 is seen in 30% cases
- Trisomy 8 and 21
- Differential diagnosis
- Aplastic anaemia and congenital marrow failure syndromes
- Acute myeloid leukaemia
- Megaloblastic anaemia
- Infections- HIV, Parvovirus, EBV, CMV, HHV6
- Toxins- Insecticides, chemotherapy, arsenic
- ALPS
- Pearson syndrome
- Radiation
- Criteria for diagnosis: Essential:
- Cytopenia involving one or more lineages
- Dysplastic changes in one or more lineages, involving at
- Blast percentage as described above
- At least 1 of following 2 criteria
- Detection of clonal cytogenetic and/or molecular abnormality
- Exclusion of other causes of cytopenia
- Non-neoplastic and some germline mutations in cases with hypocellular bone marrow
- Down syndrome, juvenile myelomonocytic leukaemia, and AML with defining genetic abnormalities in cases with hypercellular marrow
- Treatment
- HSCT is the only curative option
- With HSCT- 5year DFSR- 50%
- Myeloablative regimens are used
- No intensive chemotherapy is needed for inducing remission
Figures:
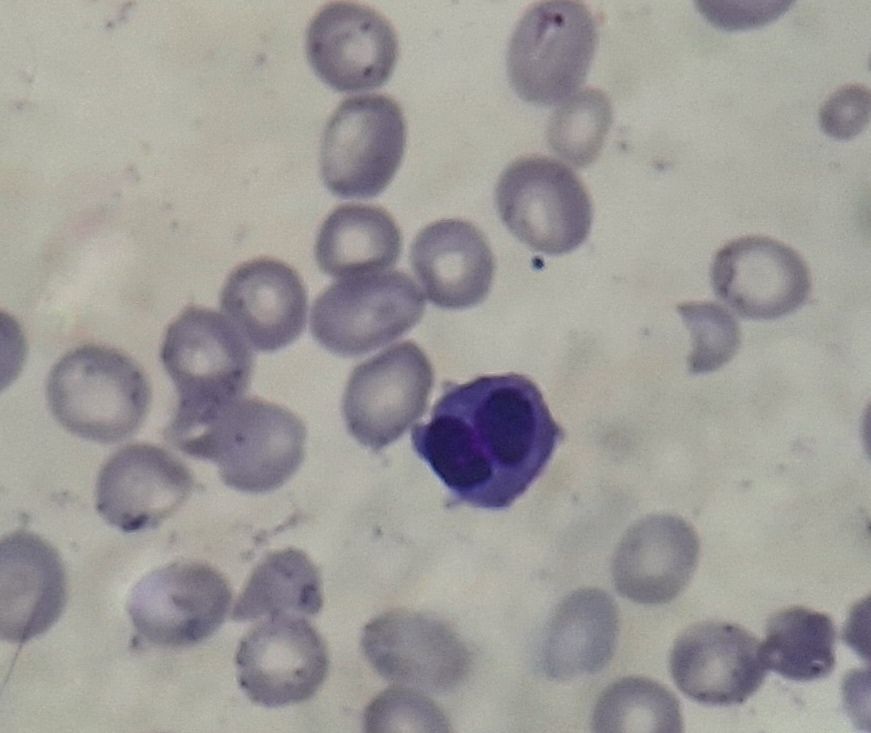
Figure 3.1.1- MDS- Dyserythropoiesis- Binucleated erythroid cell
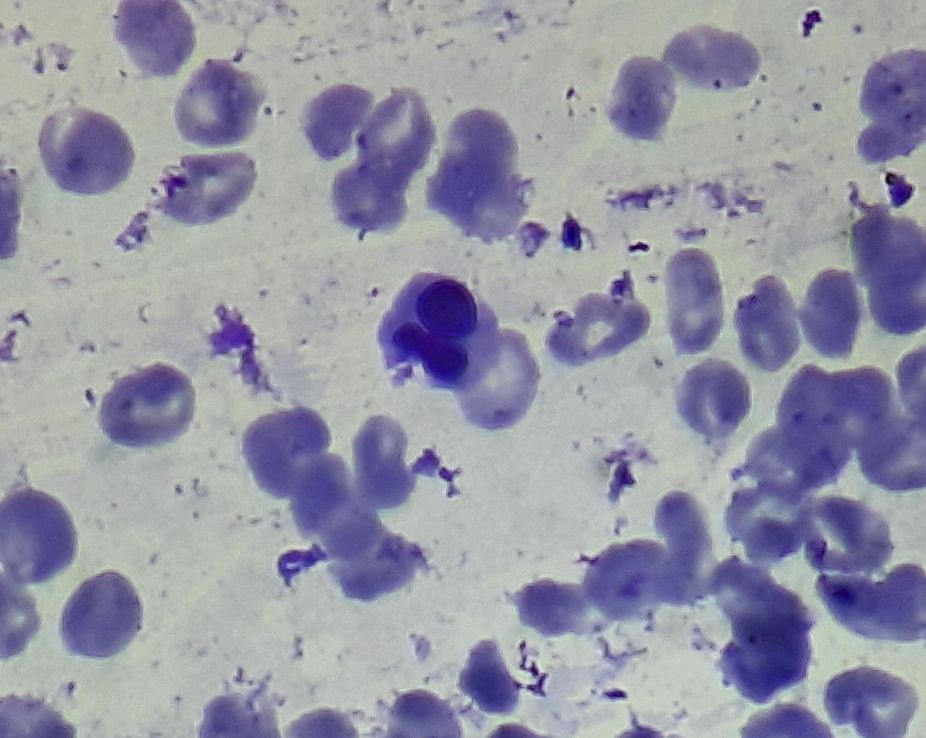
Figure 3.1.2- MDS- Dyserythropoiesis- Clover shaped nuclei
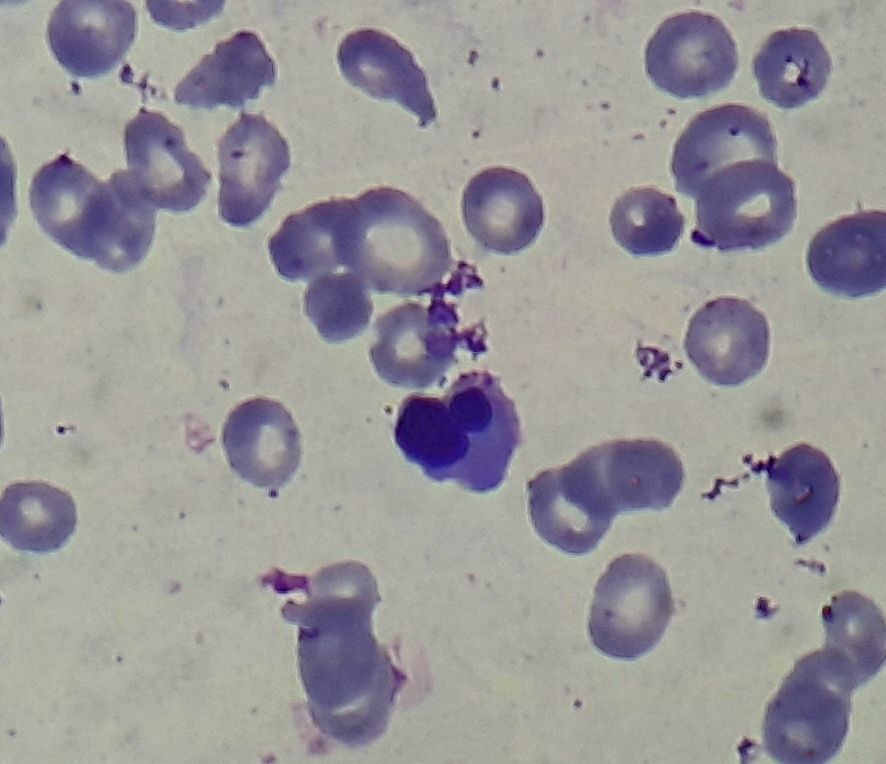
Figure 3.1.3- MDS- Dyserythropoiesis- Nuclear budding
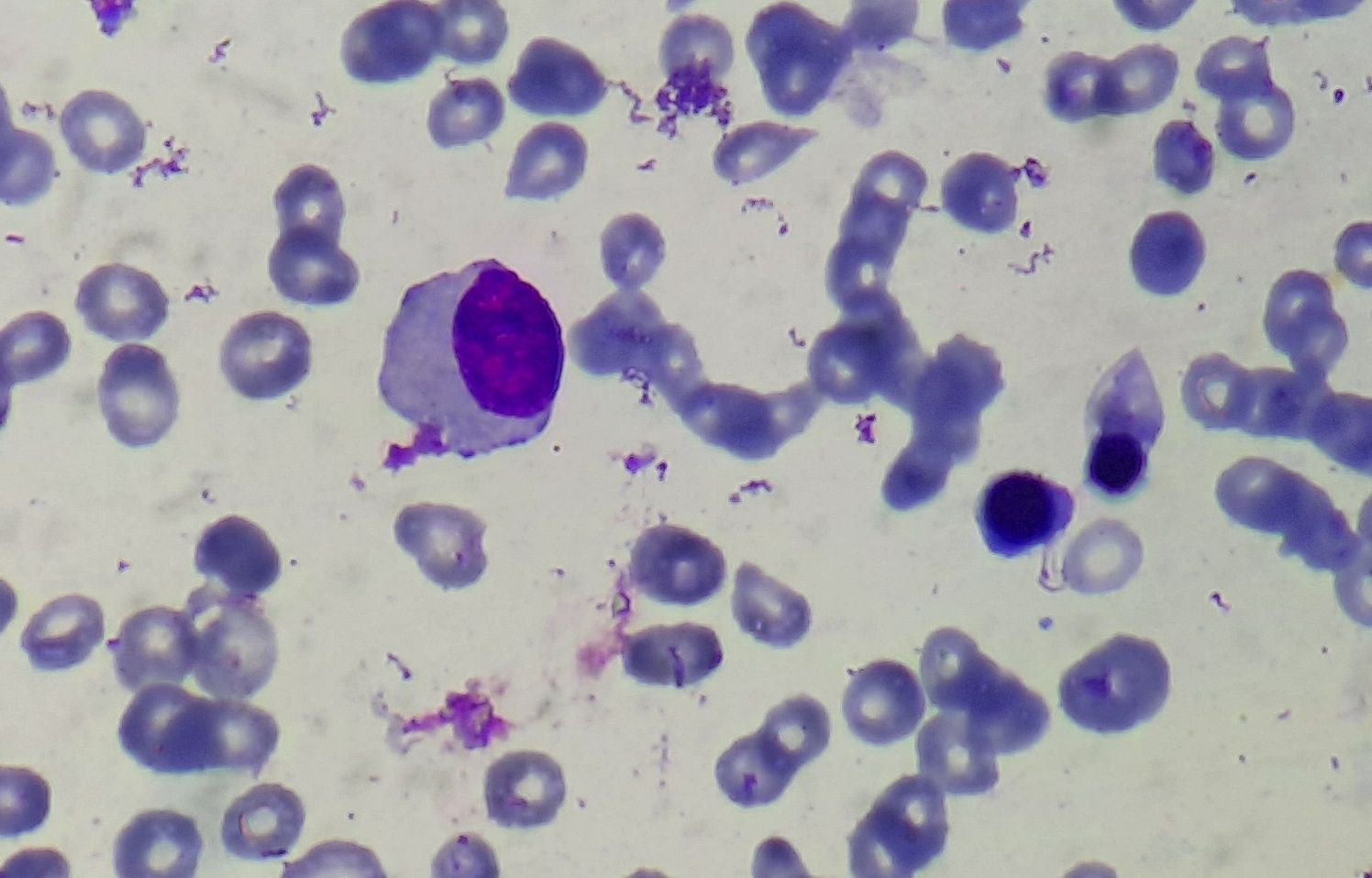
Figure 3.1.4- MDS- Dysmyelopoiesis- Hypogranular hypolobated myeloid cell
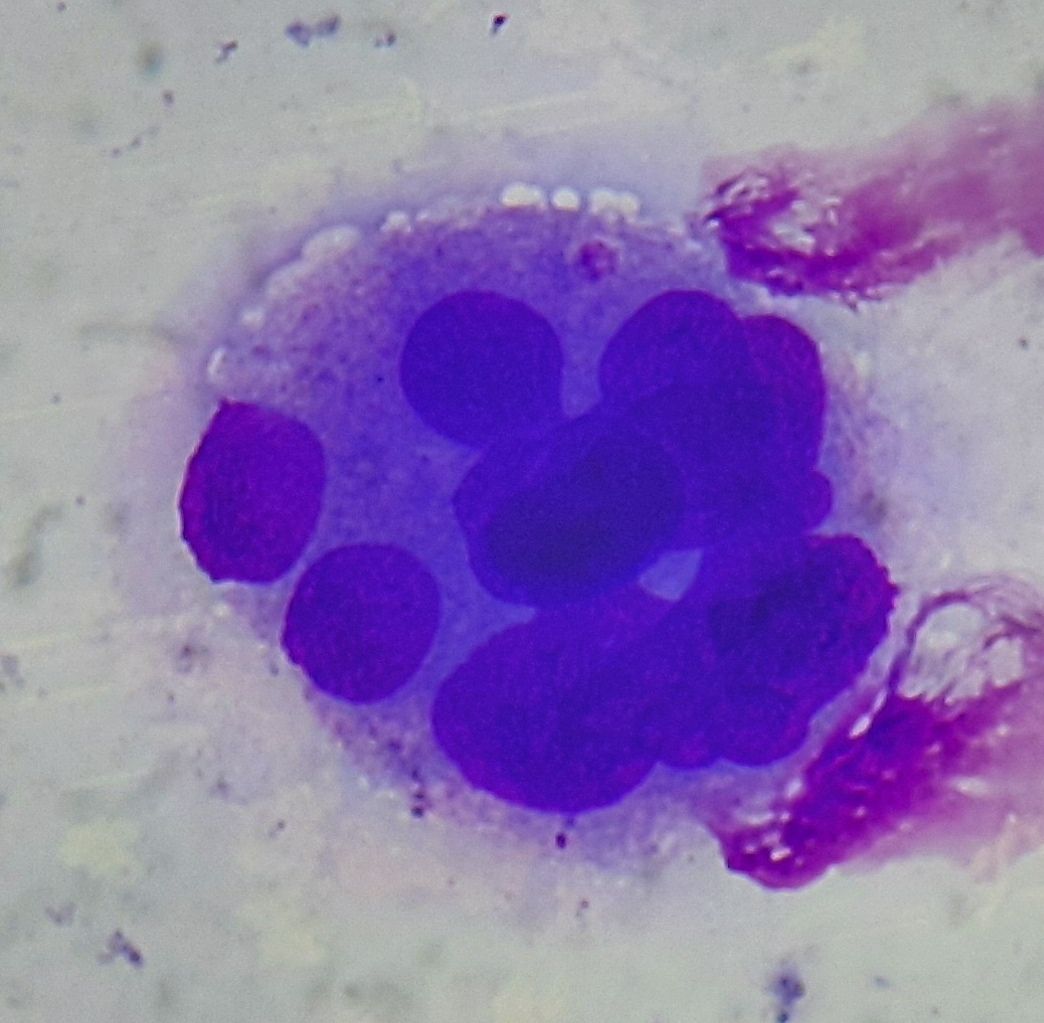
Figure 3.1.5- MDS- Dysmegakaryopoiesis
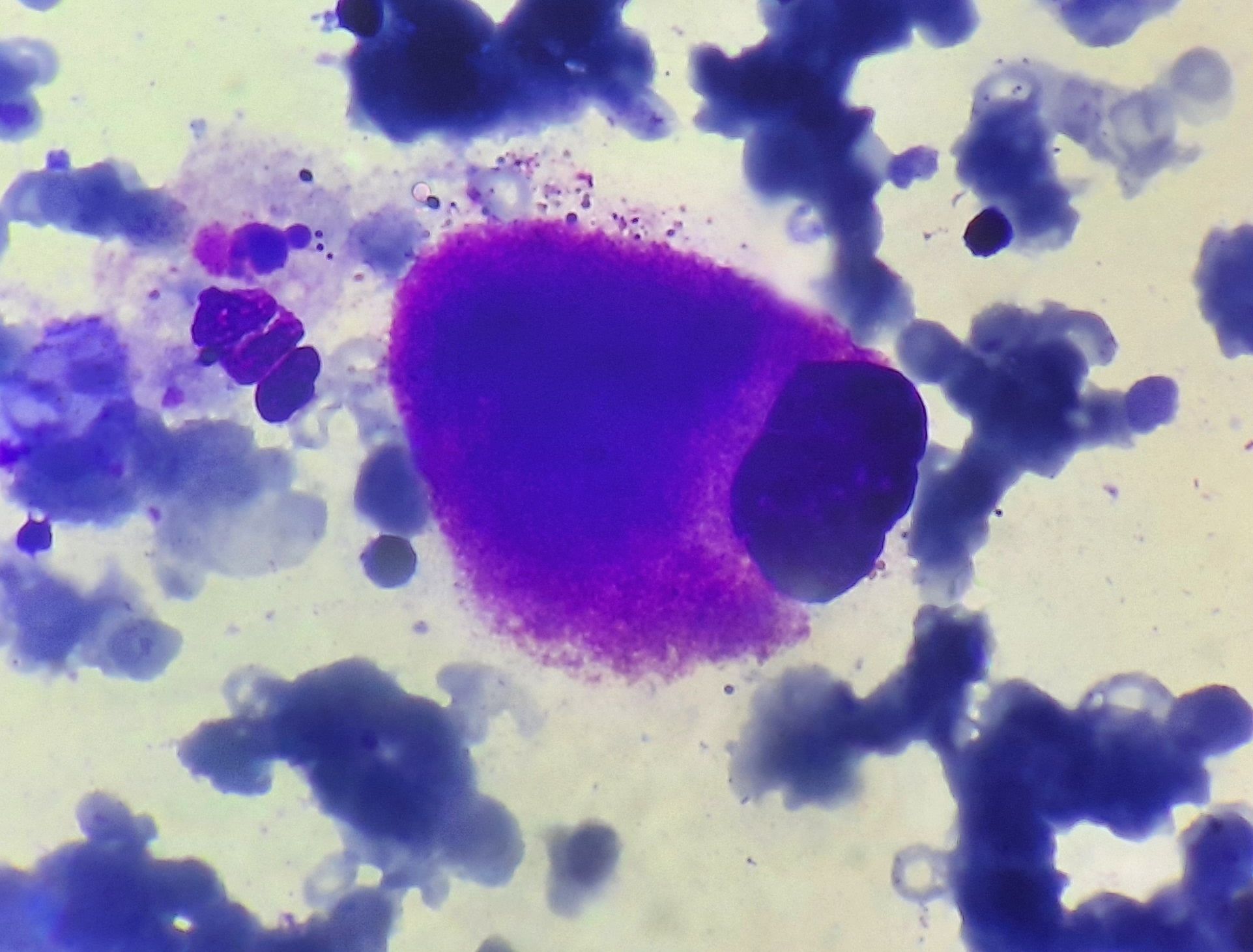
Figure 3.1.6- Megakaryocyte in 5q deletion syndrome
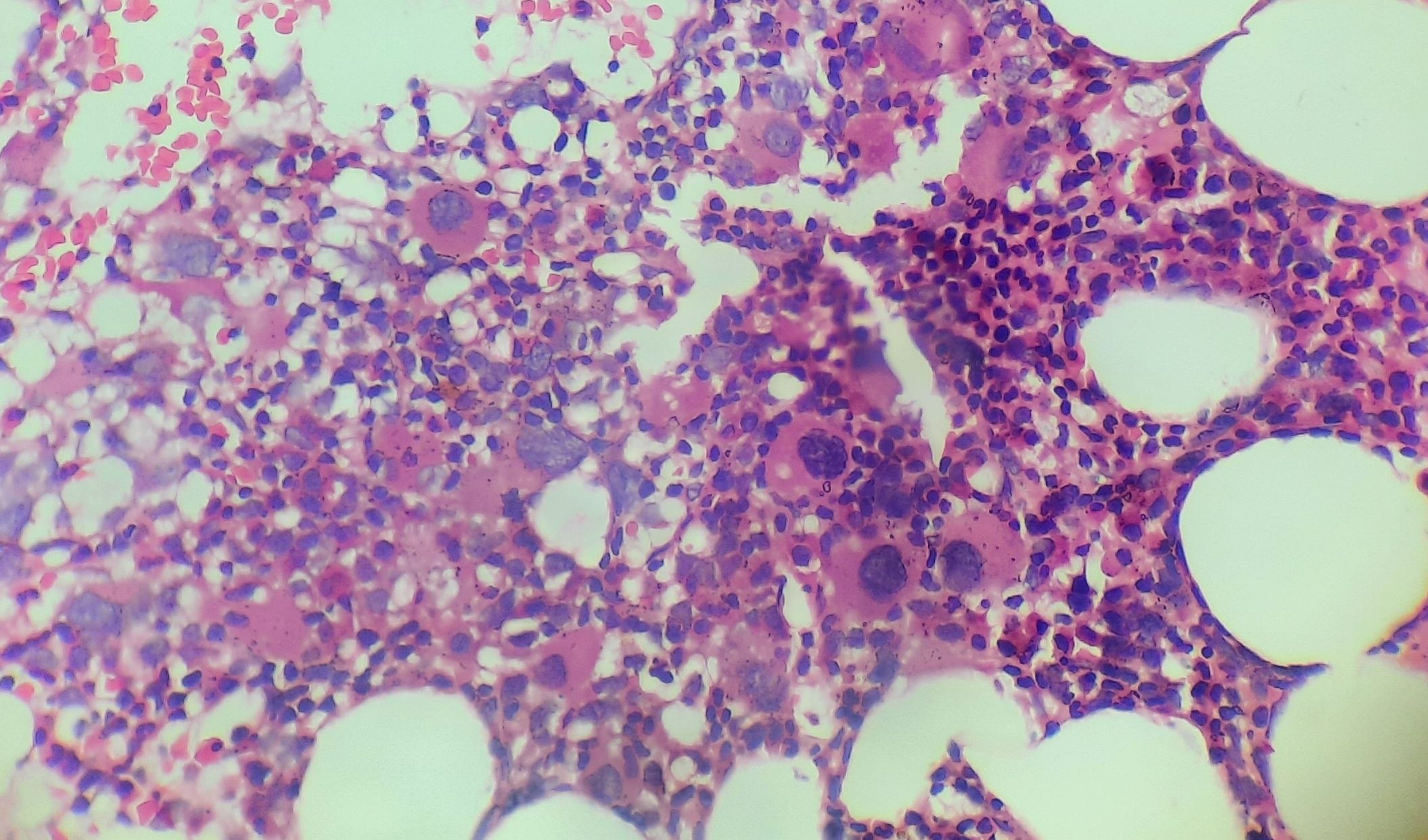
Figure 3.1.7- 5q deletion syndrome- Bone marrow biopsy
Recent advances:
Longer-term benefit of luspatercept in transfusion-dependent lower-risk myelodysplastic syndromes with ring sideroblasts
Nearly 50% of patients with lower-risk MDS require RBC transfusions within 2 years of diagnosis. Chronic RBC transfusions are associated with decreased quality of life, increased risk of iron overload, and reduced survival. In the present study, during the entire treatment phase, a significantly greater proportion of patients receiving luspatercept vs placebo achieved ≥75% reduction in RBC transfusion burden over ≥24 weeks. Luspatercept had a generally acceptable and predictable safety profile.
https://doi.org/10.1182/blood.2022016171
Eltrombopag and azacitidine in patients with high-risk myelodysplastic syndromes
In present study (the ELASTIC study) patients with baseline platelets of <150 × 109/l received eltrombopag ranging from 25 to 300 mg. Marrow response rates after three and six treatment cycles were 32% and 29% respectively. Study concluded that eltrombopag/azacitidine is safe in terms of conventional measures defined by adverse-event reporting.
https://doi.org/10.1111/bjh.18389
Lenalidomide treatment of Japanese patients with myelodysplastic syndromes with 5q deletion
Lenalidomide was approved in Japan for treating myelodysplastic syndromes with a 5q deletion (del 5q-MDS) in 2010. A post-marketing surveillance study followed 173 patients (mean age 72.4 ± 9.0 years) receiving lenalidomide between 2010 and 2011, up to 6 cycles or 6 months. Adverse reactions (ADRs) occurred in 78.0%, with common ADRs being decreased platelet and neutrophil counts, and rash. Among transfusion-dependent patients, 34.2% achieved independence. Acute myeloid leukemia (AML) progression occurred in 11.0% during the study and 17.6% in a 3-year follow-up of 68 patients.
https://doi.org/10.1007/s12185-023-03634-7
Results of EQOL-MDS trial: Eltrombopag is safe in Low-Risk Myelodysplastic Syndromes
In this multicenter trial, researchers investigated the long-term efficacy and safety of eltrombopag in low-risk MDS patients with severe thrombocytopenia. Eltrombopag showed promise, with a significant increase in platelet response compared to placebo (42.3% vs. 11.1%). Patients on eltrombopag had a 63.6% thrombocytopenia relapse-free survival at 60 months. Clinically significant bleeding was less common in the eltrombopag group. However, eltrombopag patients experienced more grade 3-4 adverse events. AML evolution and disease progression rates were similar between the eltrombopag and placebo groups.
https://doi.org/10.1200/JCO.22.02699
Risk assessment according to IPSS-M is superior to AML ELN risk classification in MDS/AML overlap patients
This study explores the classification and risk assessment of patients with Myelodysplastic Syndromes (MDS) and Acute Myeloid Leukemia (AML) overlap according to the International Consensus Classification (ICC) versus the WHO 5th edition. The findings suggest that MDS-based risk assessment using IPSS-M is applicable to MDS/AML patients, while AML-based ELN 2022 criteria are not suitable for them. MDS/AML patients classified as adverse risk by ELN 2022 had significantly longer survival than adverse risk AML patients. The study advocates for personalized risk assessment and treatment strategies based on genetic subtypes and progression markers rather than arbitrary blast cell thresholds.
https://doi.org/10.1038/s41375-023-02004-w
MDS and CMML in Japan: results of JALSG clinical observational study-11
This study conducted a multicenter, prospective observational analysis of acute myeloid leukemia (AML), myelodysplastic syndromes (MDS), and chronic myelomonocytic leukemia (CMML) in Japan, enrolling 6568 patients from August 2011 to January 2016. The report focuses on MDS (n = 2747) and CMML (n = 182) patients, with 79.5% and 79.7% aged 65 years or older, respectively. The estimated 5-year overall survival (OS) rates were 32.3% for MDS and 15.0% for CMML. Both diseases were more prevalent in men. Azacitidine was the most common treatment for MDS, used in 45.4% of higher-risk and 12.7% of lower-risk patients. The 5-year OS rate after azacitidine treatment was 12.1% for higher-risk and 33.9% for lower-risk MDS patients.
https://doi.org/10.1007/s12185-023-03686-9
Oral decitabine–cedazuridine versus intravenous decitabine for MDS and CMML.
The phase 3 trial compared oral decitabine plus cedazuridine with intravenous decitabine in patients with myelodysplastic syndromes or chronic myelomonocytic leukaemia. Results showed equivalent pharmacokinetic exposure between oral and intravenous formulations, with similar safety profiles. The most common adverse events were thrombocytopenia, neutropenia, and anaemia, with a higher incidence of serious adverse events observed with oral decitabine–cedazuridine. However, both formulations demonstrated efficacy, supporting the use of oral decitabine–cedazuridine as a safe and effective alternative to intravenous decitabine in these patients.
https://doi.org/10.1016/S2352-3026(23)00338-1
Sabatolimab plus hypomethylating agents in previously untreated patients with higher-risk myelodysplastic syndromes
Sabatolimab is an immunotherapy targeting T-cell immunoglobulin domain and mucin domain-3 expressed on immune cells and leukaemic stem cells. The STIMULUS-MDS1 trial investigated the efficacy and safety of sabatolimab plus a hypomethylating agent compared to placebo plus a hypomethylating agent in previously untreated patients with higher-risk myelodysplastic syndromes (MDS). Despite the addition of sabatolimab, there was no significant improvement in complete response rates or progression-free survival observed.
https://doi.org/10.1016/S2352-3026(23)00333-2
Prognostic impact of SF3B1 mutation and multilineage dysplasia in myelodysplastic syndromes with ring sideroblasts
This study of 170 Mayo Clinic patients found that MDS-RS-MLD had significantly worse overall survival than MDS-RS-SLD (P<0.01), but the presence of SF3B1 mutation did not impact survival. Multivariable analysis identified multilineage dysplasia, age, transfusion need, and abnormal karyotype as key prognostic factors, while SF3B1 mutations were not prognostically significant. This suggests that an MLD-based classification may be more relevant for predicting outcomes than SF3B1 mutation status in MDS-RS.
https://doi.org/10.3324/haematol.2023.284719
Luspatercept versus epoetin alfa in erythropoiesis-stimulating agent-naive, transfusion-dependent, lower-risk myelodysplastic syndromes
The COMMANDS phase 3 trial compared luspatercept and epoetin alfa in ESA-naive patients with transfusion-dependent, lower-risk myelodysplastic syndromes. Luspatercept achieved a significantly higher rate of red blood cell transfusion independence and hemoglobin improvement (60% vs 35%) compared to epoetin alfa. Both treatments had manageable safety profiles, with luspatercept showing a higher incidence of hypertension and anemia.
https://doi.org/10.1016/S2352-3026(24)00203-5
Roxadustat versus placebo for patients with lower-risk myelodysplastic syndrome
The MATTERHORN phase 3 trial evaluated roxadustat versus placebo in transfusion-dependent, lower-risk myelodysplastic syndromes patients but was terminated early due to insufficient statistical significance at interim analysis. Transfusion independence (TI) for ≥56 days occurred in 47.5% of roxadustat patients and 33.3% of placebo patients. In patients with higher transfusion burdens (≥2 pRBC units Q4W), TI rates were higher with roxadustat (36.1%) versus placebo (11.5%). Seven on-study deaths and three cases of AML progression were reported but were not treatment-related. Although the primary endpoint was not met, roxadustat showed potential in specific MDS subgroups. Further studies are warranted.
https://doi.org/10.1002/ajh.27410
An Initiative of
Veenadhare Edutech Private Limited
1299, 2nd Floor, Shanta Nivas,
Beside Hotel Swan Inn, Off J.M.Road, Shivajinagar
Pune - 411005
Maharashtra – India

howitreat.in
CIN: U85190PN2022PTC210569
Email: admin@howitreat.in
Disclaimer: Information provided on this website is only for medical education purposes and not intended as medical advice. Although authors have made every effort to provide up-to-date information, the recommendations should not be considered standard of care. Responsibility for patient care resides with the doctors on the basis of their professional license, experience, and knowledge of the individual patient. For full prescribing information, including indications, contraindications, warnings, precautions, and adverse effects, please refer to the approved product label. Neither the authors nor publisher shall be liable or responsible for any loss or adverse effects allegedly arising from any information or suggestion on this website. This website is written for use of healthcare professionals only; hence person other than healthcare workers is advised to refrain from reading the content of this website.