
howitreat.in
A user-friendly, frequently updated reference guide that aligns with international guidelines and protocols.
Primary Myelofibrosis
Updated on: 22.03.25
Introduction:
- It is a clonal myeloproliferative neoplasm characterized by proliferation of mainly megakaryocytic and granulocytic elements in bone marrow, associated with reactive deposition of bone marrow connective tissue and extra medullary erythropoiesis.
Epidemiology:
- Incidence – 0.5 – 1.5 / 1 lac population
- Commonly seen at around 70 years of age
- Equal sex incidence
- May be seen in infants with autosomal recessive inheritance
Etiology: Exact cause is not known
- Radiation
- Exposure to benzene
- Familial myelofibrosis
- Immune mechanisms- Higher incidence of MF is seen in patients with SLE
- Activating mutations of JAK/STAT pathway
- JAK2V617F is seen in 65% of cases. Exon 12 mutation is infrequent.
- CALR mutation- Seen in 25-35% patients
- MPL mutation- Seen in 5-10% cases
- Triple negative primary myelofibrosis: 8-10% cases
- Clonal driver mutations:
- Ten-eleven translocation 2 (TET-2)- 4q24- Seen in 17% cases
- Enhancer of Zeste homolog 2 (EZH2)- 7q36.1- Seen in 13% cases
- Casitas B Lineage Lymphoma (CBL)- 11q 23.3 Exons 8 and 9- Seen in 6% cases
- Isocitratedehydrogenase (IDH)- 1 and 2- 2q33.3/15q 26.1- Seen in 4% cases
- Ikaros Family Zinc Finger 1 (IKZF1)- 7p12- Infrequent
- Additional sex combs like 1 (ASXL1)- 20q11- Infrequent
- Other mutations include: DNMT3A, SRSF2, N-RAS, K-RAS, CBL and TP53
Pathogenesis:
1.
Hyperactivation of thrombopietin/MPL/JAK2 axis
and
Down-regulation of GATA1 expression
↓
Defective package of growth factors such as PDGF, Fibroblast growth factor, MMP9 and TGF beta in neoplastic megakaryocytes
↓
Inappropriate release of these growth factors
↓
These factors diffuse into intercellular milieu
↓
Proliferation of fibroblasts and neovascularization due to proliferation of capillaries
Fibroblasts are not clonal; hence they are not tumor cells. But hematopoietic cells are.
2.
Epigenetic Methylation of CXCR4 promoters
↓
Decreased CXCR4 mRNA
↓
Decreased CXCR4 expression on CD34+ cells
↓
Enhanced migration of CD34+ cells in blood (These cells have predisposition to differentiate into megakaryocytes)
3.
Increase in circulating endothelial cell progenitors + Release of EGFbeta, BFGF and VEGF from megakaryocytes
↓
Increased angiogenesis
4.
Over-expression of FKBP51 on megakaryocytes
↓
Resistance to apoptosis through calcineurin pathway
5.
Increased number of CD34+ cells exit from BM
↓
Filtered in spleen due to abnormal trafficking pattern
↓
Accumulate progressively and continue to proliferate
↓
Extramedullary hematopoiesis (myeloid metaplasia) and splenomegaly
- Functional abnormalities of neutrophils: Impaired phagocytosis, oxygen consumption, nitro blue tetrazolium reduction, hydrogen peroxide generation, decreased myeloperoxidase.
- Platelet abnormalities: Impaired aggregation with epinephrine, depletion of dense granules adenosine diphosphatase content, decreased platelet lipo-oxygenase pathway
Clinical Features:
- Prefibrotic phase- Asymptomatic without any organomegaly. CBC may show mild anemia, mild leucocytosis and/or thrombocytosis. (Close differential diagnosis is ET. Refer ET for BM findings in these conditions. Follow up usually clarifies the diagnosis)
- Fibrotic phase:
- Hypercatabolic state: Low grade fever, night sweats, fatigue, weight loss, Pruritus
- Anemia: Occurs due to
- Ineffective erythropoiesis
- Inflammatory iron sequestration from elevated hepcidin
- Splenic sequestration
- Autoimmune hemolysis
- Myelosuppression from medications
- Bleeding from portal hypertension
- Splenomegaly – Massive. Often leads to early satiety.
- Hepatomegaly – Slight to moderate, firm, smooth non tender
- Bleeding manifestations due to thrombocytopenia
- Thromboembolism- Venous/ arterial
- Bone pain
Complications:
- Bone marrow failure leading to frequent infections and hemorrhage
- Portal hypertension (Non-cirrhotic) due to
- Extramedullary hematopoiesis in liver
- Massive increase in hepatic blood flow and intrahepatic obstruction.
- Perisinusoidal fibrosis
- Collagen bundles in space of Disse
- Cardiac failure due to anemia
- Leukemic transformation – Seen in 20-25% patients
- Development of PNH clone
- Acquired HbH disease
- Pure red cell aplasia
- Pulmonary hypertension
- Nephrotic syndrome
- Mass of extramedually hematopoiesis- Pulmonary, GI, CNS, genitourinary
- Neutrophilic dermatosis (Sweet's syndrome)- which can progress to bullae/ pyodermagangrenosum
- Leukemia cutis- Larger cells are seen which are CD61 positive
Investigations:
- Hemogram
- Prefibrotic phase:
- No or mild leukoerythroblastosis
- No or minimal red blood cell poikilocytosis; few if any dacrocytes
- Mild anemia
- Mild to moderate leucocytosis
- Mild to marked thrombocytosis
- Fibrotic phase
- Normocytic normochromic anemia, prominent red blood cell poikilocytosis with dacrocytes (Tear drop cells)
- Leucocytosis with TLC usually between 10,000-14,000/cmm, with <10% blasts/ Leucopenia
- Leucoerythroblastic blood picture- Plenty of nRBCs with shift to left in myeloids up to blasts
- Slight basophilia
- Thrombocytosis/ thrombocytopenia.
- Giant platelets with abnormal platelet granulation.
- Pancytopenia is noted in 10% of cases.
- Prefibrotic phase:
- Bone Marrow :
- Prefibrotic phase
- Hypercellularity
- Neutrophilic proliferation
- Megakaryocytic proliferation and atypia (clustering of megakaryocytes, hyperchromatic and abnormally lobulated or cloud like megakaryocytic nuclei, naked megakaryocytic nuclei)
- Minimal or absent reticulin fibrosis. If present it tends to be concentrated around blood vessels.
- Erythropoiesis is reduced
- Fibrotic phase:
- Aspirate is generally a dry tap.
- Increased cellularity- Hypercellular fragments and hypocellular trails
- Biopsy shows extensive fibrosis
- Dilated marrow sinuses with intraluminal hematopoiesis
- Prominent megakaryocytic proliferation and atypia (clustering of megakaryocytes, abnormally lobulated megakaryocytic nuclei. Dense chromatin clumping, nuclear hyperchromasia, bulbous/ cloud like nuclear lobes and naked nuclei are seen)
- Increased number of dysmorphic megakaryocytes are seen, even in fibrotic areas.
- Increased pathologic emperipolesis- Entry of neutrophils and other marrow cells into canalicular system of megakaryocyte. (This is also responsible for alfa granule injury and release of TGF beta and PDGF)
- Erythropoiesis- Suppressed
- Granulocytic hyperplasia
- Reticulin and/or collagen fibrosis of varying degrees is seen (Usually MF-2 to MF-3)
- New bone formation (osteosclerosis)
- Prefibrotic phase
Grading of fibrosis
Grading | Description |
MF-0 | Scattered linear reticulin with no intersections (cross-overs). Corresponds to normal bone marrow |
MF-1 | Loose network of reticulin with many intersections, especially in perivascular areas |
MF-2 | Diffuse and dense increase in reticulin with extensive intersections, occasionally with focal bundles of collagen and/or focal osteosclerosis |
MF- 3 | Diffuse and dense increase in reticulin with extensive intersections and coarse bundles of collagen, often associated with osteosclerosis |
Fibrotic stroma is rich in Type 1 and 3 collagen. Reticulin stain principally stains type 3 collagen. Thick fibers with type 1 collagen stain with trichrome stain. Hence this stain is positive only in advanced disease.
Accelerated phase of PMF - 10-19% blasts seen in blood or marrow
Blast phase of PMF - ≥20% blasts in blood or bone marrow
- Cytogenetics –
- Nothing specific.
- Seen in 45% patients.
- Common abnormalities: del (13q) , del (20q), partial trisomy 1q, +8, +9
- Abnormalities associated with poor prognosis: Complex karyotype, inv (3), monosomy 5 or del (5q), monosomy 7 or del (7q), del 12p, 11q23 rearrangement, and i(17q)
- Molecular tests for myeloproliferative neoplasm related mutations- BCR-ABL1, JAK 2, CAL R and MPL.
- Myeloid mutation panel:
- SRSF2, ASXL1, EZH2, IDH1 and U2AF1-Q157 mutations predict inferior survival (hence called high-molecular-risk mutations)
- RAS/CBL mutations predict resistance to ruxolitinib
- Type 1/like CALR mutation is associated with superior survival.
- ESR-Elevated
- Neutrophil alkaline phosphatase score-Raised
- Serum uric acid level-Raised
- Serum LDH level – Raised
- Serum folate level – Decreased (Due to utilization by abnormal tissue)
- X-Ray of bone
- Patchy sclerosis of medullary cavity
- Coarsening of trabeculation
- Rarefaction giving mottled appearance
- MRI
- New bone formation and periosteal thickening
- Altered hyperintensity of T1 weighted images that normally result from marrow fat
- Lumbar spine dual energy X ray absorption and quantitative CT
- Increased bone formation
- Bone thickening
- Increased cancellous and woven bone
- Rarely osteolytic lesions
- Immunophenotyping - Nothing specific
- Number of circulating CD34 cells is increased
- Immunohistochemistry
- CD34 and CD117: help in enumerating number of blasts
- CD61 and CD42b: Highlight megakaryocytes
- CAL2 is positive in megakaryocytes if CALR is mutated.
Criteria for Diagnosis:
Prefibrotic/Early stage:
Requires that all 3 major criteria and at least 1 minor criterion (Minor criteria must be met which is confirmed in 2 consecutive determinations).
- Major criteria
- Megakaryocytic proliferation and atypia, without reticulin fibrosis/ grade 1 fibrosis accompanied by increased age-adjusted bone marrow cellularity, granulocytic proliferation, and decreased erythropoiesis
- WHO criteria for CML/PV/ET/MDS not met
- Positive for JAK 2/CALR/MPL
Or
presence of another clonal marker (By cytogenetics or NGS panel for myeloid neoplasms)
Or
Absence of minor reactive bone marrow reticulin fibrosis (Due to conditions such as infection, autoimmune disorder or other chronic inflammatory conditions, hairy cell leukaemia or another lymphoid neoplasm, metastatic malignancy)
- Minor criteria:
- Anaemia not attributed to a co-morbid condition
- Leucocytosis- >11,000/cmm
- Palpable splenomegaly
- LDH level above the upper limit of the institutional reference range
Overt Fibrotic stage:
All 3 major and at least 1 minor criteria (Minor criteria must be met which is confirmed in 2 consecutive determinations)
- Major:
- Megakaryocytic proliferation and atypia, accompanied by reticulin fibrosis grades 2 or 3
- WHO criteria for CML/PV/ET/MDS not met
- Positive for JAK 2/CALR/MPL or presence of another clonal marker such as ASXL1, EZH2, TET2, IDH1, IDH2, SRSF2 and SF3B1 (By cytogenetics or NGS panel for myeloid neoplasms) or absence of reactive myelofibrosis
- Minor:
- Anemia not attributable to comorbid condition
- Leucocytosis- >11,000/cmm
- Palpable splenomegaly
- Raised LDH
- Leukoerythroblastosis
Prognosis:
- Overall mean survival is 3-5 years
International Prognostic Scoring System (IPSS)
Prognostic variable | 0 Point | 1 Point |
Age in years | ≤65 | >65 |
WBC count (/cmm) | ≤25,000 | >25,000 |
Hemoglobin (gm/dL) | ≥ 10 | <10 |
Peripheral blood blasts (%) | <1 | ≥1 |
Constitutional symptoms (>10% weight loss in 6 months, night sweats, unexplained fever higher than 37.5°C) | Absent | Present |
Risk Group | Points | Median survival |
Low | 0 | 135 months |
Intermediate 1 | 1 | 95 months |
Intermediate 2 | 2 | 48 months |
High | ≥ 3 | 27 months |
Dynamic International Prognostic Scoring System (DIPSS)
Prognostic variable | 0 Point | 1 Point | 2 points |
Age in years | ≤65 | >65 |
|
WBC count (/cmm) | ≤25000 | >25000 |
|
Hemoglobin (gm/dL) | ≥ 10 |
| <10 |
Peripheral blood blasts (%) | <1 | ≥1 |
|
Constitutional symptoms | Absent | Present |
|
Risk Group | Points |
Low | 0 |
Intermediate 1 | 1 or 2 |
Intermediate 2 | 3 or 4 |
High | 5 or 6 |
DIPSS-PLUS Scoring system
Prognostic variable | Points |
DIPSS- Low risk | 0 |
DIPSS- Int-1 | 1 |
DIPSS- Int- 2 | 2 |
DIPSS- High risk | 3 |
Platelets <1lac/cmm | 1 |
Transfusion need | 1 |
Unfavorablekaryotype* | 1 |
* Unfavorablekaryotypes include: Complex karyotype (>2 abnormalities) or trisomy 8, 7/7q-, i(17q), 5/5q-, 12p-, inv(3) or 11q23 rearrangements
Risk Group | Points |
Low | 0 |
Intermediate 1 | 1 |
Intermediate 2 | 2 or 3 |
High | 4 to 6 |
MIPSS70+ v2.0 is a prognostic scoring system
Prognostic variable | 0 Point | 1 Point | 2 points | 3 points | 4 points |
Karyotype* | Other karyotype | Unfavorable karyotype | VHR karyotype | ||
HMR mutations (ASXL1, SRSF2, EZH2, IDH1, IDH2, U2AF1 Q157) | No HMR mutations | 1 HMR mutation | ≥2 HMR mutations |
| |
Absence of type 1/like CALR mutation | No | Yes |
|
| |
Presence of constitutional symptoms | No | Yes |
|
| |
Anemia severity | Mild or no | 8 to 9.9 in women and 9 to 10.9 in men | <8 in women and <9 in men |
|
|
≥2% circulating blasts | No | Yes |
|
|
* Cytogenetics:
- Very high risk (VHR): Single/multiple abnormalities of -7, i(17q), inv(3)/3q21, 12p-/12p11.2, 11q-/11q23, or other autosomal trisomies not including + 8/ + 9 (eg, +21, +19).
- Favorable: Normal karyotype or sole abnormalities of 13q-, +9, 20q-, chromosome 1 translocation/duplication or sex chromosome abnormality including -Y.
- Unfavorable: All other abnormalities.
Points | Risk group | 10-year overall survival | Median overall survival (years) |
0 | Very low risk | 86% | Not reached |
1 to 2 | Low risk | 50% | 10.3 |
3 to 4 | Intermediate risk | 30% | 7 |
5 to 8 | High risk | 10% | 3.5 |
9 to 14 | Very high risk | <3% | 1.8 |
RR6 model
- Predicts survival in MF based on clinical response after 6 months of ruxolitinib
- Takes into account: Spleen size below costal margin, Ruxolitinib dose and RBC transfusions done (at diagnosis, at 3 months and at 6 months)
- Link for calculating RR6 score: http://www.rr6.eu/
Myelofibrosis transplant scoring system (MTSS)
- Predict survival after allo-HSCT in transplant-eligible patients
Prognostic variable | Points |
Age ≥57 y | 1 |
PS- Karnofsky <90% | 1 |
Mismatched unrelated donor | 2 |
WBC >25, 000/cmm | 1 |
PLT <1,50,000/cmm | 1 |
Absence of CALR/MPL | 2 |
ASXL1 mutation | 1 |
Score | Risk Category | 5 year survival |
0-2 | Low | 90% |
3-4 | Int | 77% |
5 | High | 50% |
6-9 | Very high | 34% |
MYSEC-PM score:
- Used specifically for patients with post-PV MF and post-ET MF
- Calculation:
- Age: 0.15 × y of age points
- Constitutional symptoms: 1 point
- Hb <11 g/dL: 2 points
- Blasts ≥3%: 2 points
- PLT <150000/cmm: 1 point
- Absence of CALR: 2 points
Score | Risk Category | Median survival |
<11 | Low | Not reached |
11-13 | Int-1 | 9.3 y |
14-15 | Int-2 | 4.4 y |
≥16 | High | 2 y |
Differential diagnosis:
- Other causes of myelofibrosis: Refer Pancytopenia section (Click Here)
Pretreatment Work-up:
- History : Symptom burden (MPN-SAF- Total Symptom Score 10): Click here
- Examination
- Spleen (cm below costal margin):
- WHO P. S.
- BSA
- BMA and Bx
- Reticulin/ Trichrome stain
- Hemoglobin
- TLC, DLC
- Platelet count
- Peripheral smear
- LFT- Bili- T/D SGPT: SGOT: Albumin: Globulin:
- Creatinine
- Uric acid
- Electrolytes : Na: K: Ca: Mg: PO4:
- LDH
- HIV
- HBsAg
- HCV
- Cytogenetics
- BCR-ABL1
- JAK 2
- CAL-R
- MPL-1
- Myeloid mutation panel (Must include: ASXL1, CBL, CSF3R, DNMT3A, EZH2, KIT, KRAS, IDH1/2, NRAS, RUNX1, SETBP1, SF3B1, SH2B3, SRSF2, TET2, TP53 and U2AF1)
- USG Abdomen with portal venous Doppler
- Oesophagogastroduodenoscopy if signs of portal HTN present: To rule out occult varices
- HLA Typing for transplant candidate
- Prognostic score
- Chemotherapy consent after informing about disease, prognosis, cost of therapy, side effects, hygiene, food and contraception
- Tumor board meeting and decision
- Attach supportive care drug sheet
- Inform primary care physician
Treatment Plan:
Low risk patients with low platelet counts or complex cytogenetics may be evaluated for Allo-SCT.
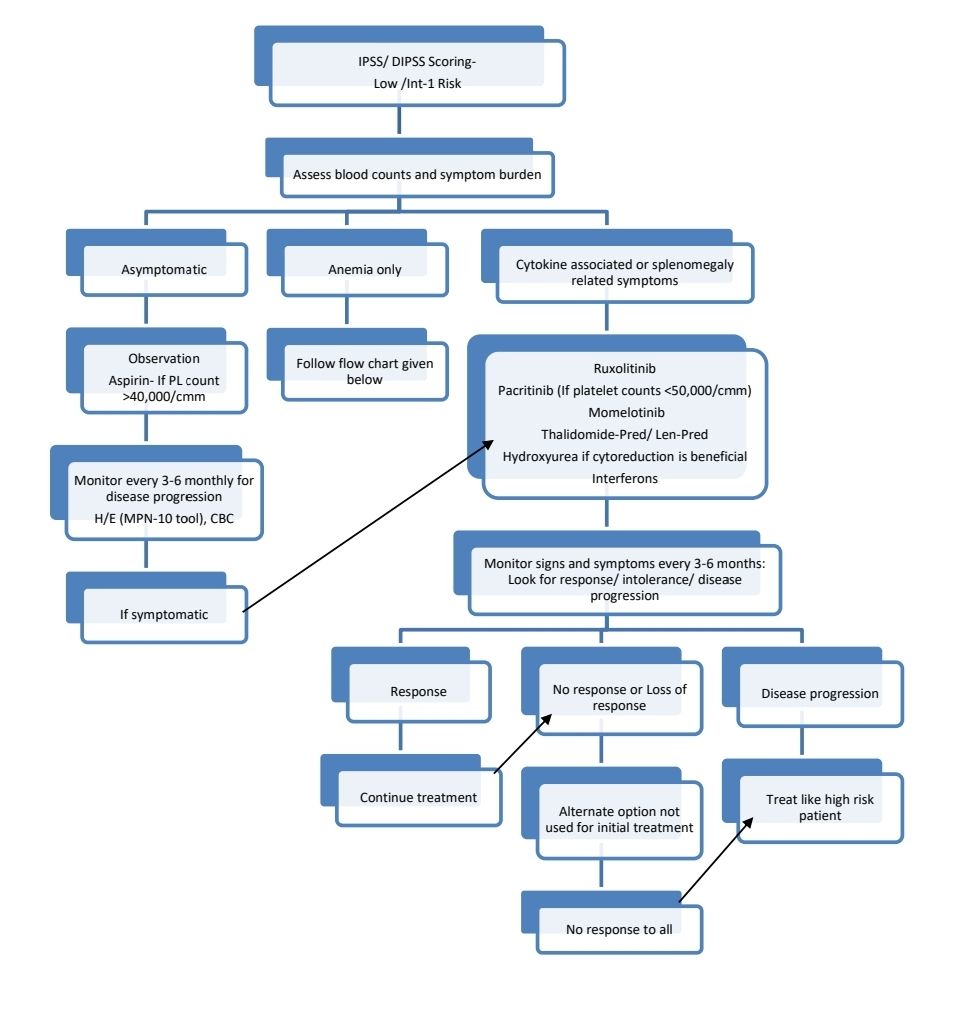
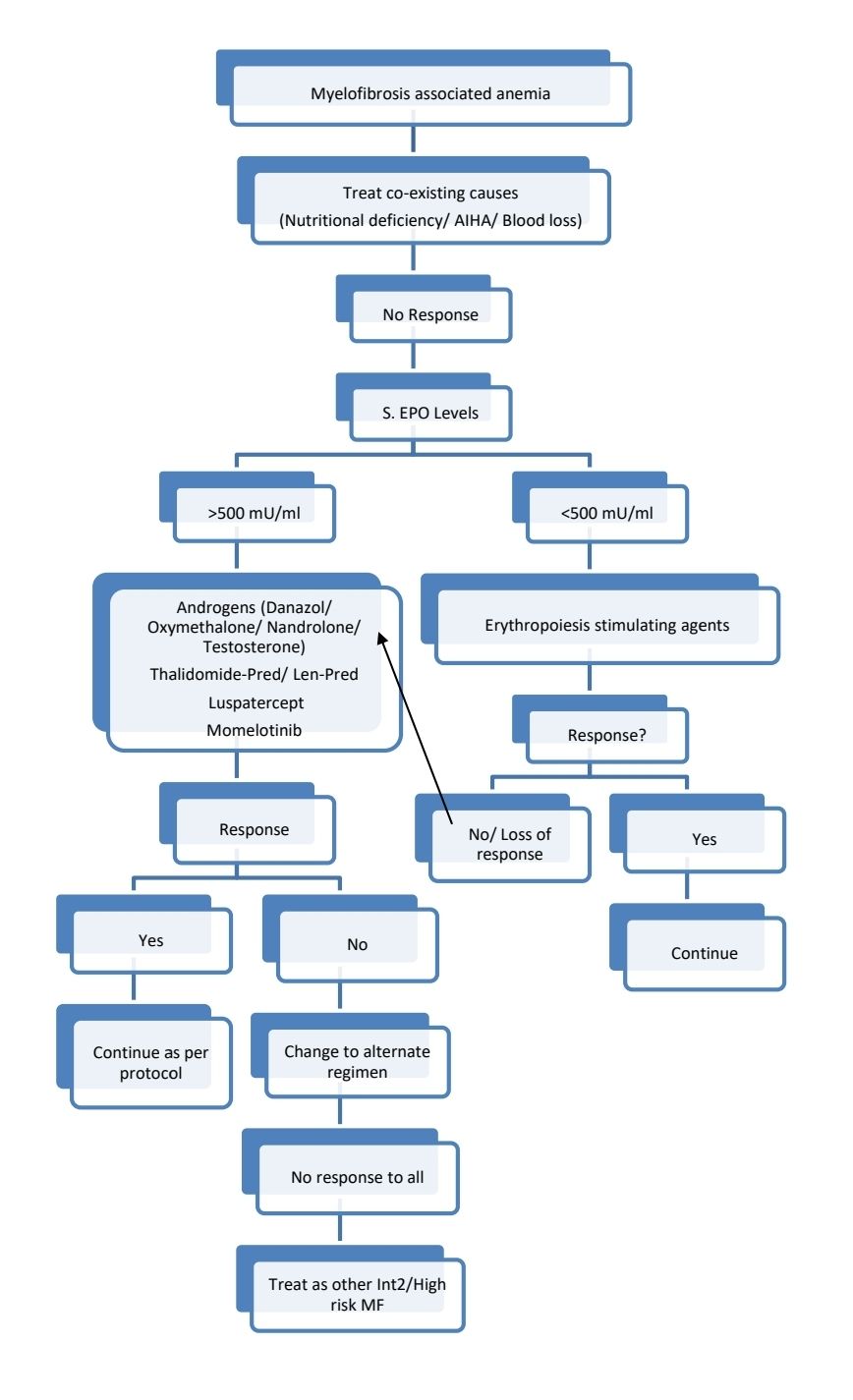
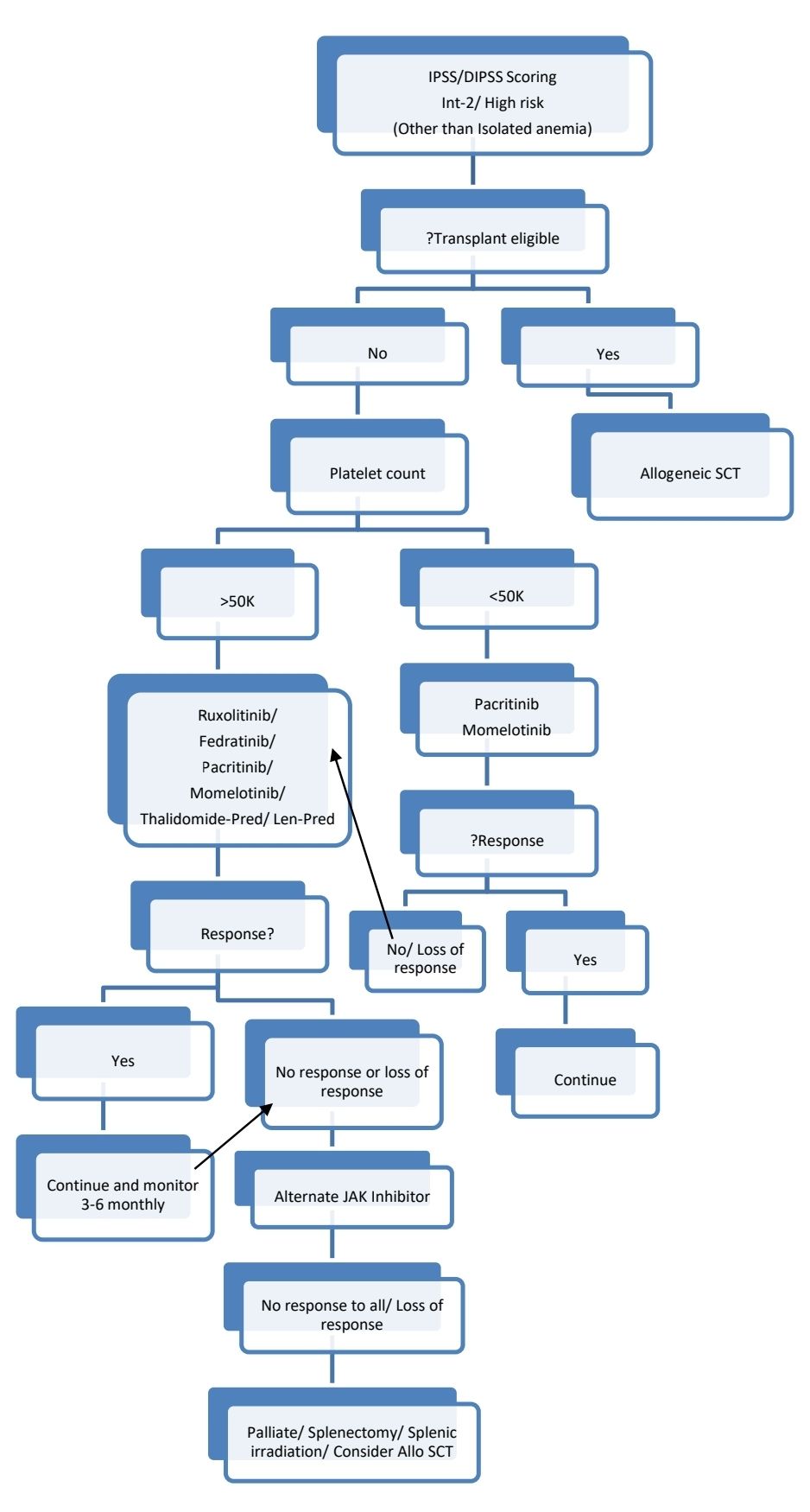
Myelofibrosis with accelerated phase/ MF with blast phase
- Median survival- <1year
- Transplant candidate: Induce remission with hypomethylating agents/ intensive chemotherapy (such as 7+3 induction). This should be followed by allogeneic SCT in CR1.
- Not a transplant candidate:
- Hypomethylating agents- Azacytidine, Decitabine with/without JAK inhibitors
- Low intensity induction chemotherapy
Response Criteria (Benefit must last for >12 weeks to qualify as response):
Complete response:
- Bone marrow- Normocellular for age with <5% blasts and ≤ Grade 1 fibrosis
- Peripheral blood- Hb>10gm/dL, ANC >1000/cmm, Platelet count >1lac/cmm, <2% immature myeloid cells (Includes blasts, promyelocytes, myelocytes and metamyelocytes)
- Complete resolution of symptoms and non palpable spleen and liver.
Partial response:
- Same as above but one of either of BM or Peripheral blood criteria are fulfilled along with clinical criteria.
For Ruxolitinib, to assess symptom improvement, Myeloproliferative Neoplasm Symptom Assessment Form is used, which is available here.
DIPSS score must be calculated at least once a year.
About Each Modality of Treatment:
- Ruxolitinib
- COMFORT-I trial
- Primary targets: JAK1/2
- Dose: 15mg- BD. If there is pre-existing anemia/ thrombocytopenia, start with a dose of 5mg- BD.
- It is useful even in patients without JAK 2 mutation.
- Avoid in patients with platelet counts <50,000/cmm.
- Hold if there is worsening of anemia, thrombocytopenia. Restart with lower dose and use EPO/Darbapoietin and/or danazol.
- Other side effects include: headache, bruising, dizziness, diarrhea, weight gain and hypercholesterolemia.
- Useful in reducing splenomegaly and and myelofibrosis related symptoms such as pruritis.
- Shown to decrease hepatomegaly and portal hypertensionwhich occurs because of myelofibrosis.
- High risk of HBV reactivation. Hence HBV testing and prophylaxis if required must be given.
- There is high risk of reactivation of tuberculosis, hence take appropriate measures.
- Stop if there is no benefit after 3 months or side effects are not tolerable.
- Taper the dose over 10 days while stopping with coverage of steroids, as abrupt stopping causes reaction resembling systemic inflammatory response syndrome
- Fedratinib:
- JAKARTA and FREEDOM trials
- Primary targets: JAK2, JAK1, TYK2, JAK3
- Dose: 400mg- OD
- Useful in reducing splenomegaly and constitutional symptoms
- Used in Intermediate 2 and High risk patients.
- Similar to Ruxolitinib, can cause worsening of anemia and thrombocytopenia.
- Other side effects include: nausea, diarrhea
- Better to do thiamine level measurement/ supplementation as Wernicke’s encephalopathy incidence is high.
- Pacritinib
- PERSIST trial
- Dose: 400mg- OD
- Primary targets: JAK2, FLT3, IRAK1, CSF1R, ACVR1
- Side effects: anemia, thrombocytopenia, diarrhea, cardiac events
- Momelotinib
- SIMPLIFY and MOMENTUM trials
- Dose: 200mg- OD
- Significantly reduces RBC transfusions
- Primary targets: JAK1, JAK2, ACVR1
- Side effects: Anemia, thrombocytopenia
- Thalidomide/Lenalidomide with prednisolone
- Thalidomide- 50mg- OD or Lenalidomide 10mg- OD for 3 months
- Then taper Prednisolone-
- 0.5 mg/kg/day- first month
- 0.25 mg/kg/day- Second month
- 0.125 mg/kg/day- Third month
- If response after 3 months of therapy- Continue thalidomide for next 3 months
- Presence of del 5q is associated with better response rates with lenalidomide
- Hydroxyurea
- Dose: Up to 1gm/day
- Compared to other MPN, marrow tolerance to hydroxyurea is low
- Given for cytoreduction, if there is leukocytosis or thrombocytosis
- Helps to decrease constitutional symptoms and organomegaly as well
- Benefit is seen in 8-10 weeks of treatment
- Erythropoiesis stimulating agents
- Inj. Erythropoietin- 10,000 units- 3 times a week
- Inj. Darbepoetin- 150microgm- weekly
- Androgens -
- Danazol – 400 – 600 mg/day or Oxymetholone – 200 mg – oral – OD or testosterone enanthate 400–600 mg IM weekly or oral fluoxymesterone 10 mg- TID
- Help to increase erythropoiesis
- 1/3 rd patients respond
- Continue for minimum of 6 months before evaluating response
- Continue for 6 more months in those patients who respond, then titrate the dose to minimum to maintain response
- Screen for prostate cancer in men
- Monitor LFT
- Interferon-
- It is the only available treatment that can act on the clonal disorder, possibly by inducing an immune response against the malignant cells
- Very less clinical effect
- Pegylated IFN is better.
- Luspatercept:
- Dose: 1.0-1.75 mg/kg, 21-day cycles
- Common side effect: Hypertension
- Allogeneic bone marrow transplant:
- Only therapy with a chance of cure
- Indications:
- Intermediate 2 and High risk patients
- Intermediate 1 patients with low platelet counts and complex cytogenetics
- NGS showing "higher risk" mutations
- Consider following before selecting patient for BMT:
- Age
- Performance score
- Major co-morbid conditions
- Psychosocial status
- Patient preference
- Availability of care giver
- If patient is in blast crisis, bridging therapy is necessary to decrease the blast percentage to acceptable levels
- Challenges unique to PMF include:
- Advanced age and poor PS of patients
- Malnutrition from constitutional symptoms
- Associated problems such as splenomegaly, pulmonary hypertension and portal hypertension
- Abnormal/ fibrotic marrow microenvironment
- Splenectomy prior to transplant leads to earlier engraftment, without impact on overall survival or graft failure. Hence not recommended routinely.
- Better results in patients receiving<20 blood transfusions
- Usually RIC regimens are used.
- JAK inhibitors have to be given for at least 2 months prior to transplant to improve general condition and decrease splenemegaly. This must be done even if patient is already transplant eligible. JAK inhibitors can be tapered prior to or during conditioning. Tapering must be completed before stem cell infusion.
- If >10% blasts are present in peripheral blood, Azacytidine can be added to JAK inhibitors, to decrease blast count.
- 5 year overall survival-30%- 68%
- TRM at 1 year- 16-48%
- Post transplant monitoring- JAK2 PCR. Molecular relapse to be treated with donor lymphocyte infusions.
Supportive Care:
- Supportive PRBC and platelet transfusions
- Use leucoreduced products in prospective transplant candidate
- Tranexamic acid, if bleeding is refractory to transfusions
- Iron chelation after 20 transfusions
- Prophylactic antibiotics if there are recurrent infections. G-CSF should be used with caution, due to risk of splenic rupture
- Folic acid 5mg- OD
- Aspirin- 75mg- OD - If there is thrombocytosis
- Allopurinol if there is hyperuricemia
- Masses of extramedullary hematopoiesis: Local radiation
- Severe bone pain-
- Etidronate- 6mg/kg/day on alternate months
- Radiation can be used
- Portosystemic shunt surgery:
- If there is gastro-esophageal variceal bleeding or if there is refractory ascites
- If portal hypertension is due to increased flow from spleen to liver- Splenectomy is the treatment of choice
- If portal hypertension is due to intrahepatic block or hepatic vein thrombosis- Splenorenal shunt or TIPSS is the treatment of choice.
Other Treatment Options:
- Low dose Melphalan- 2.5mg- Thrice a week
- Splenectomy:
- High risk of serious peri-operative complications such as bleeding, thrombosis and infections
- It is difficult as spleen is adherent to neighboring serosal surfaces and organs
- With best centers morbidity and mortality are 31% and 9% respectively
- Indications include severe thrombocytopenia and very high transfusion requirements who have failed JAK2 inhibitor therapy.
- Laparoscopic splenectomy is not advised
- Long term penicillin prophylaxis has to be given
- Splenic irradiation:
- Useful especially in case of painful splenomegaly
- Platelet count must be >50,000/cmm prior to radiation
- Can lead to severe cytopenia
- Used for patients who are not fit for splenectomy
- Etanercept
- 25mg- SC- Twice weekly
- Associated with improvement in constitutional symptoms such as weight loss, night sweats, fatigue, fever etc
- Cyclosporine
- Achieve trough levels of 100-200ng/ml
- Useful in patients with immune disorders (DCT/ ANA positive patients)
Therapies presently under trial:
- Jaktinib
- Dose: 100mg-BD
- Primary targets: JAK2, JAK1, ACVR1, TYK2
- Side effects: Anemia and thrombocytopenia
- Itacitinib
- Dose: 100mg- BD
- Common side effects: Anemia, thrombocytopenia, Fatigue
- Combination therapies:
- Ruxolitinib combined with pelabresib (oral BET inhibitor), navitoclax (oral BCL-XL/BCL-2 inhibitor), and parsaclisib (PI3Kδ inhibitor)
Special Situations:
- Pregnancy:
- Similar to management of essential thrombocythemia with pregnancy
- Children:
- Following must be ruled out before making diagnosis of PMF
- Acute panmyelosis with myelofibrosis
- Acute megakaryocytic leukemia
- Autoimmune disorders, NK cell proliferations
- Familial cases of infantile myelofibrosis
- Causes of secondary myelofibrosis such as rickets
- Hypocellular MDS
- Many cases presenting in infancy eventually "burn out" with spontaneous erythropoietic recovery, occurring as early as 2-3 years after diagnosis
- Curative option for "true" PMF- Allo SCT
- Trial of steroids should be considered, once AMKL and rickets have been excluded.
- Following must be ruled out before making diagnosis of PMF
Autoimmune myelofibrosis:
- 8 diagnostic criteria must be met
- Grade 3 or 4 reticulin fibrosis
- Lack of clustered or atypical megakaryocytes
- Lack of dysplasia/ eosinophilia/ basophilia
- Lymphoid infiltration of marrow
- Lack of osteosclerosis
- Absent or mild splenomegaly
- Presence of autoantibodies
- Lack of disorders associated with PMF
Figures:
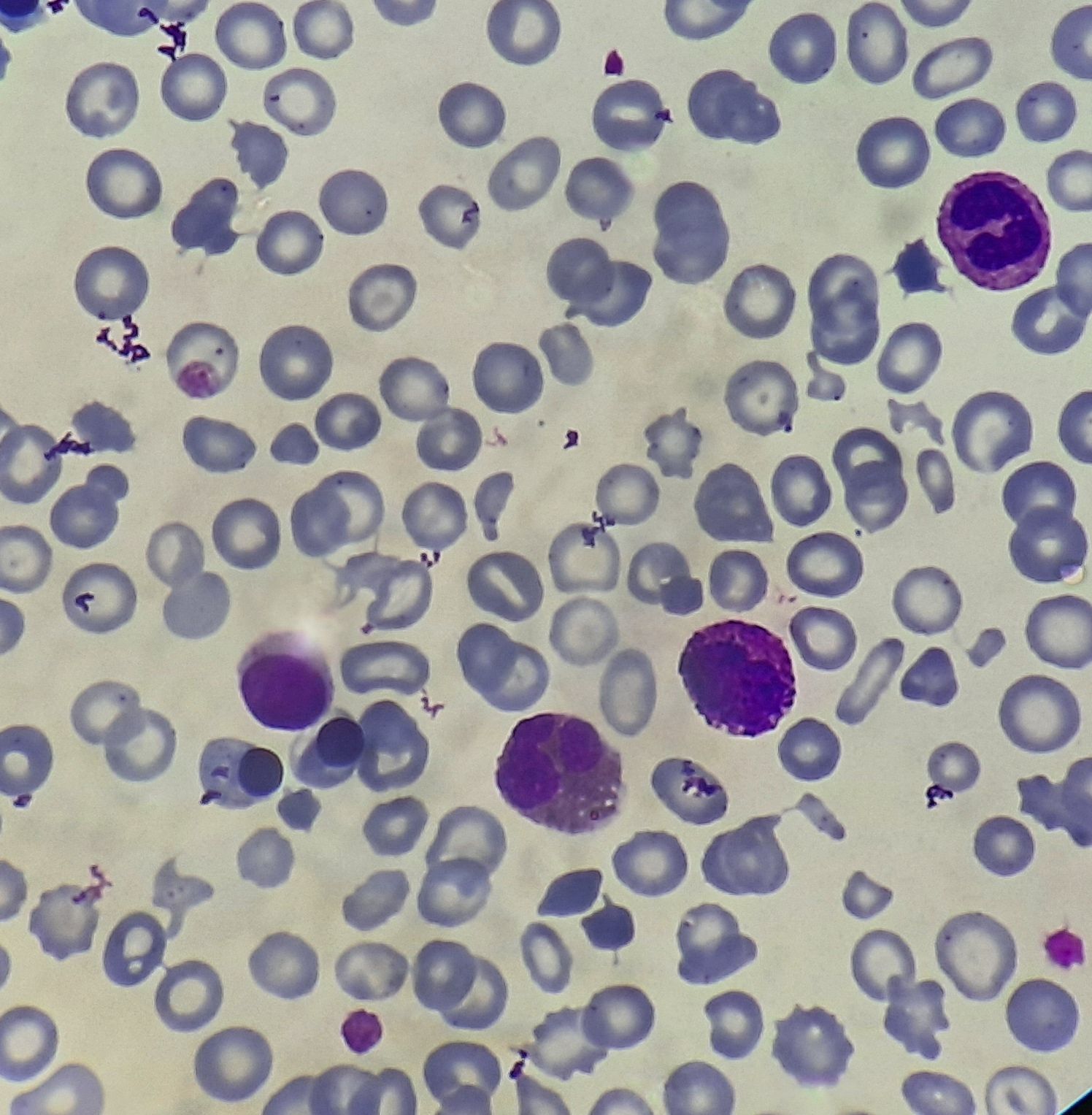
Primary myelofibrosis- Prefibrotic phase- Peripheral smear
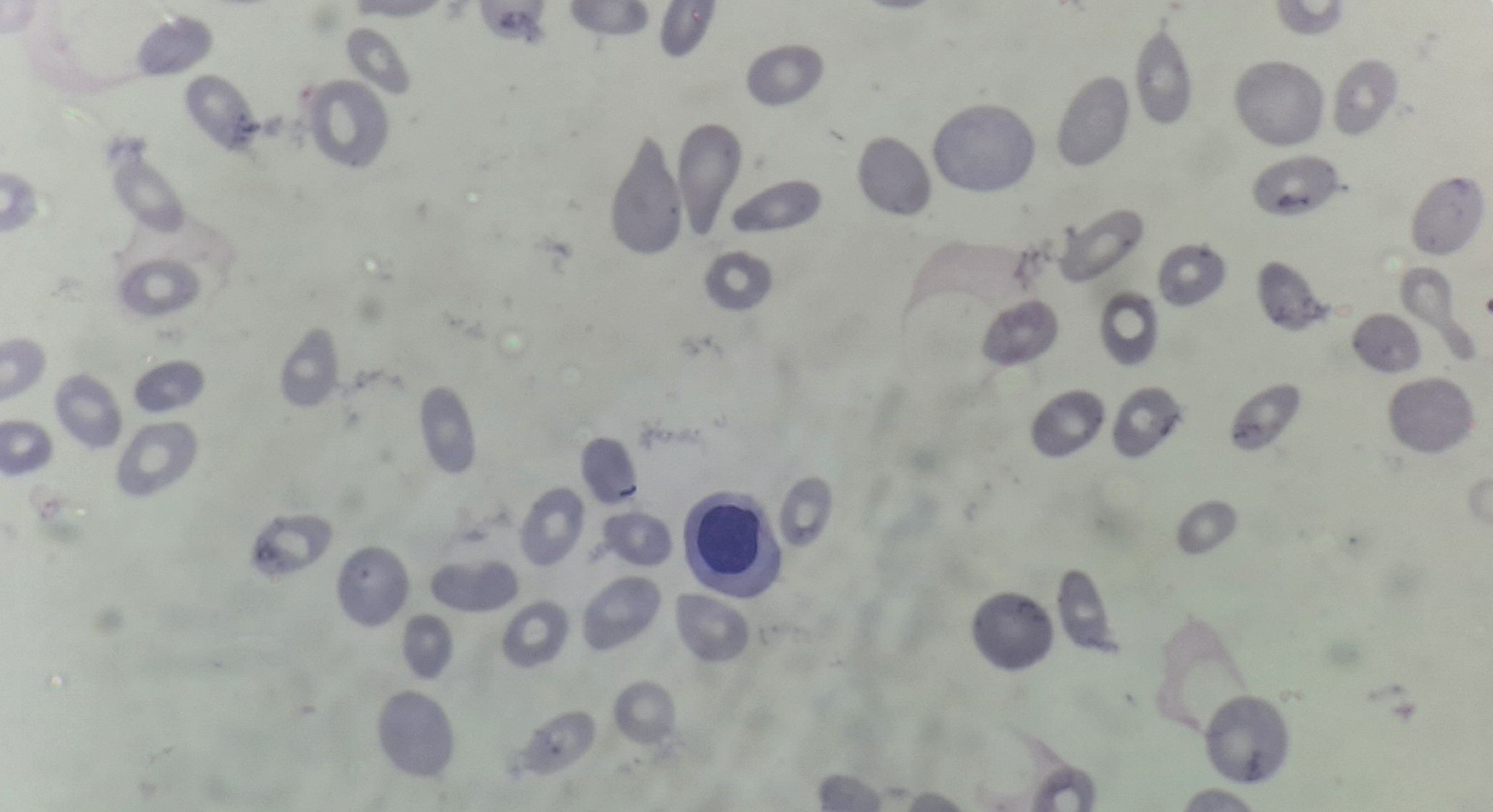
Primary myelofibrosis- Fibrotic phase- Peripheral smear
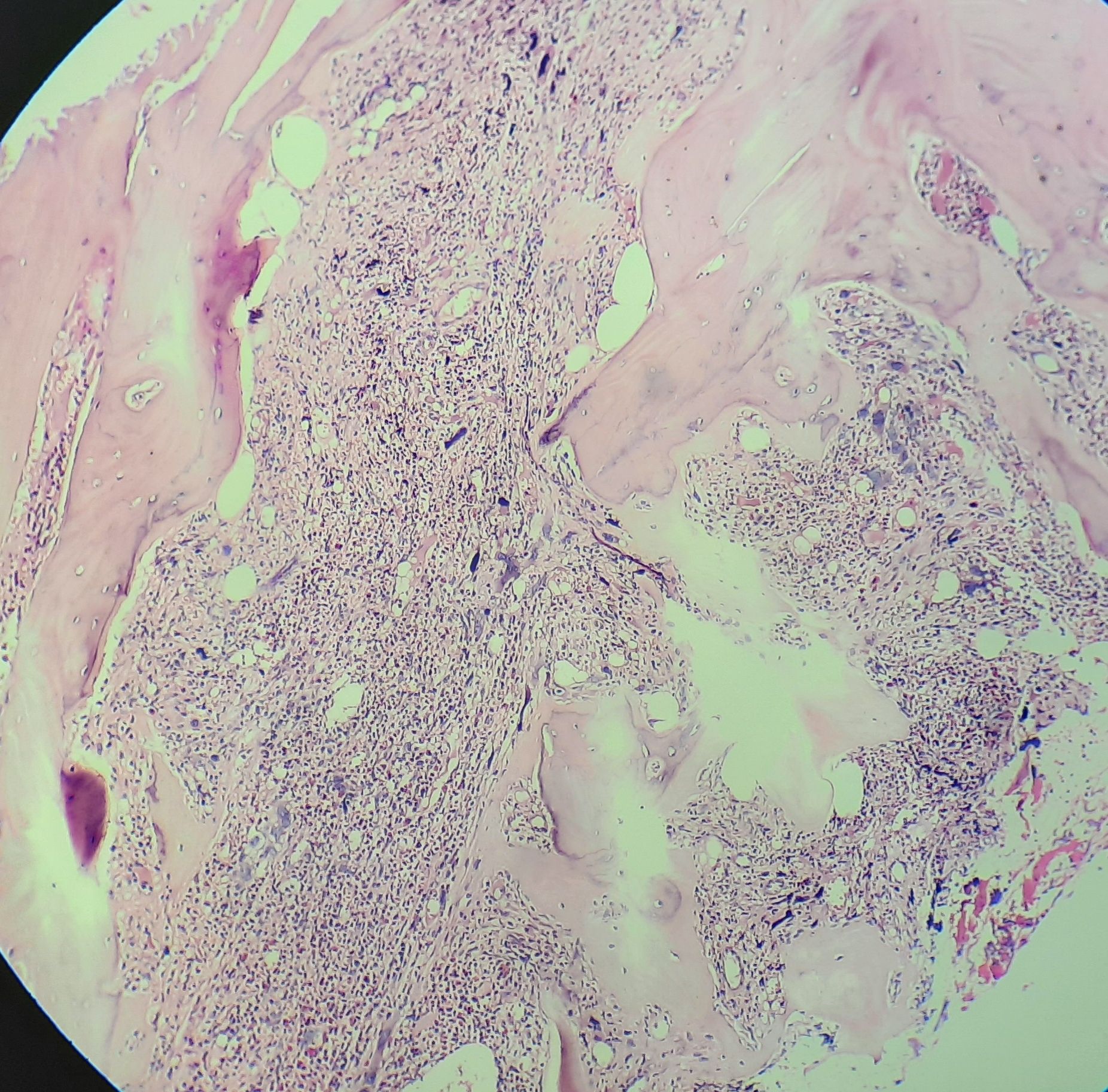
Primary myelofibrosis- Fibrotic phase- Bone marrow biopsy
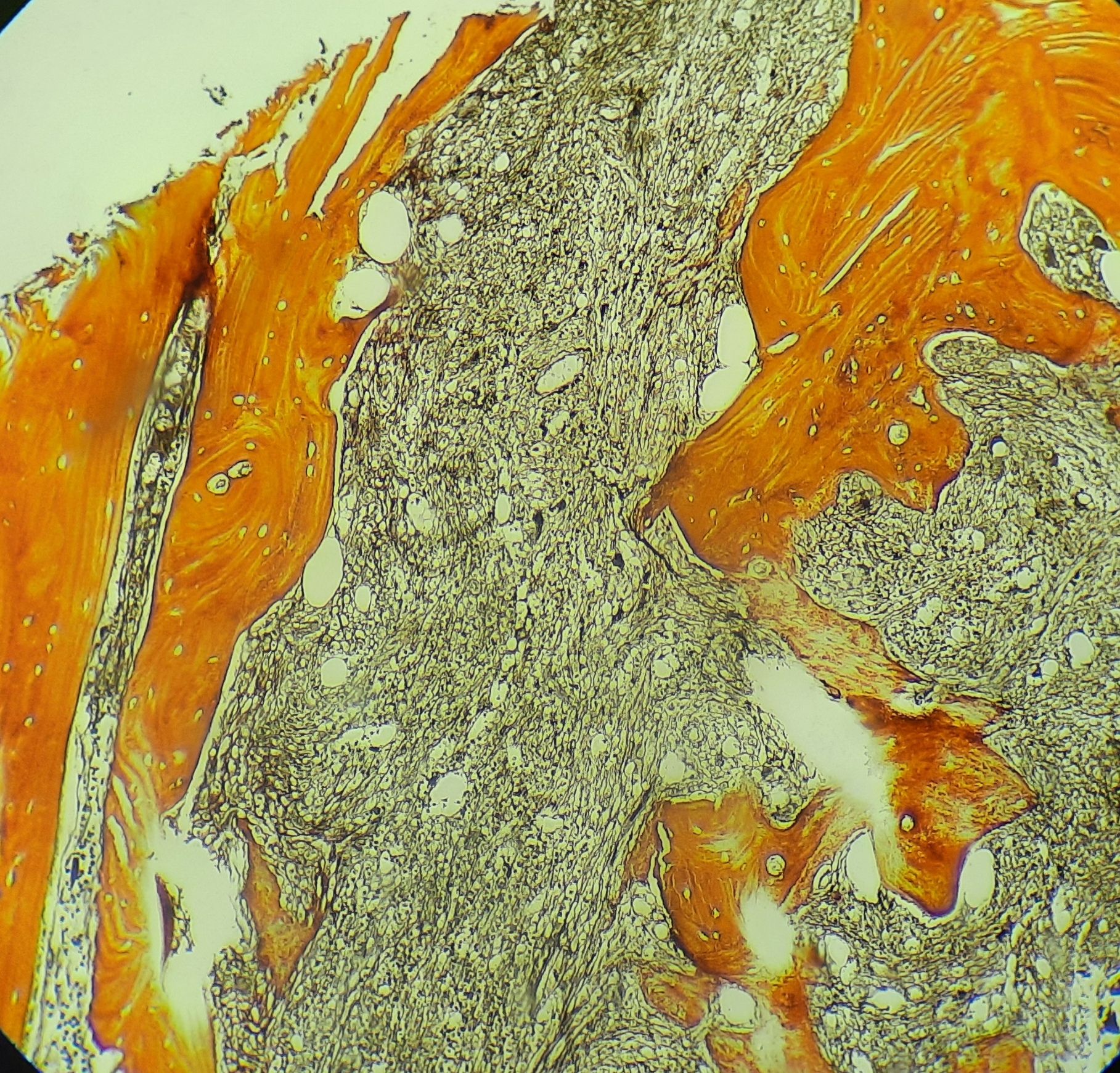
Primary myelofibrosis- Fibrotic phase- Bone marrow biopsy- Reticulin stain
Recent advances:
Addition of Navitoclax to Ruxolitinib improves overall outcome in patients with primary myelofibrosis
Navitoclax is a BCL-XL/BCL-2 inhibitor. This drug was evaluated in high risk myelofibrosis patients who had suboptimal response to ruxolitinib. There was significant decrease in splenic size, decrease in total symptom score and improvement of hemoglobin levels. Follow-up bone marrow biopsy showed significant decrease in BM fibrosis.
https://doi.org/10.1200/JCO.21.02188
Zinpentraxin alfa in myelofibrosis
Zinpentraxin alfa, a recombinant form of PTX-2, an antifibrotic protein, was studied in a phase II trial involving patients with myelofibrosis (MF). The trial included 27 patients who received zinpentraxin alfa either alone or with ruxolitinib. The primary endpoint, the overall response rate (ORR), was 33% at week 24. Some patients also experienced improvements in symptom scores and bone marrow fibrosis. Most adverse events were mild, with fatigue being the most common. Anemia and thrombocytopenia were infrequent, and serious treatment-related adverse events occurred in a small number of patients. Zinpentraxin alfa showed promise as a potential therapy for MF in this trial.
https://doi.org/10.3324/haematol.2022.282411
Pelabresib in Combination With Ruxolitinib in Naïve Myelofibrosis
In a phase II study (MANIFEST), the combination of the bromodomain and extraterminal domain inhibitor (BETi) pelabresib with ruxolitinib in JAK inhibitor-naïve patients with myelofibrosis showed promising results. At 24 weeks, 68% of patients achieved a spleen volume reduction of ≥ 35%, and 56% achieved a total symptom score reduction of ≥ 50%. Additional benefits included improved hemoglobin levels, reduced fibrosis, and a reduction in JAK2V617F-mutant allele fraction. The combination therapy was generally well-tolerated, with thrombocytopenia and anemia being the most common grade 3 or 4 toxicities.
https://doi.org/10.1200/JCO.22.01972
Upfront allogeneic transplantation versus JAK inhibitor therapy for patients with myelofibrosis
The study aimed to compare outcomes of patients aged 70 or below with myelofibrosis (MF) in chronic phase who received upfront JAK inhibitor (JAKi) therapy versus upfront allogeneic hematopoietic cell transplantation (HCT) in dynamic international prognostic scoring system (DIPSS)-stratified categories. The results showed that, for the entire cohort, median overall survival (OS) was longer for patients who received JAKi compared to upfront HCT. In patients with intermediate-2 and high-risk disease, median OS was not significantly different between JAKi and HCT. The study suggests that a universal upfront HCT approach for higher-risk MF may not provide significant benefits.
https://doi.org/10.1038/s41409-023-02146-6
Allogeneic hematopoietic cell transplantation in patients with CALR-mutated myelofibrosis
The study analyzed outcomes of 346 CALR-mutated myelofibrosis (MF) patients who underwent allogeneic hematopoietic cell transplantation (allo-HCT) in 123 EBMT centers between 2005 and 2019. After a median follow-up of 40 months, the estimated overall survival (OS) rates at 1, 3, and 5 years were 81%, 71%, and 63%, respectively. Patients receiving busulfan-containing regimens achieved a 5-year OS rate of 71%. Non-relapse mortality (NRM) at 1, 3, and 5 years was 16%, 22%, and 26%, respectively, while the incidence of relapse/progression was 11%, 15%, and 17%, respectively. Older age correlated with worse OS, while primary MF and HLA-mismatched transplants had a near-to-significant trend to decreased OS. Comparative analysis between CALR- and JAK2-mutated MF patients revealed better OS, lower NRM, lower relapse, and improved graft-versus-host disease-free and relapse-free survival in CALR-mutated patients.
https://doi.org/10.1038/s41409-023-02094-1
Splenic irradiation for myelofibrosis prior to hematopoietic cell transplantation
This study investigated the safety and efficacy of splenic irradiation before allogeneic hematopoietic cell transplantation (HCT) in patients with myelofibrosis who failed Janus kinase (JAK) inhibition. Among 59 patients, splenic irradiation led to significant spleen size reduction in 97% of cases, with a median decrease of 5.0 cm. The 3-year overall survival rate was 62%, and the 1-year non-relapse mortality was 26%. Splenic irradiation, when adjusted for confounders, was associated with significantly reduced relapse rates compared to immediate HCT or splenectomy, demonstrating its potential benefit in this patient population.
https://doi.org/10.1002/ajh.27252
Momelotinib versus ruxolitinib in JAK inhibitor-naïve patients with myelofibrosis
This sub-analysis of SIMPLIFY-1 compared momelotinib and ruxolitinib in JAK inhibitor-naïve Japanese myelofibrosis patients. At 24 weeks, momelotinib showed a 50% spleen response rate (SRR) versus 44.4% with ruxolitinib. Total symptom score response was 33.3% for momelotinib and 0% for ruxolitinib, while transfusion independence rates were 83.3% and 44.4%, respectively. Momelotinib was well tolerated and reduced transfusion needs, with fewer grade 3/4 adverse events than ruxolitinib.
https://doi.org/10.1007/s12185-024-03822-z
Treatment of myelofibrosis with refractory anemia with luspatercept
In this retrospective study, 18 patients with refractory anemic myelofibrosis were treated with luspatercept for at least 9 weeks. Erythroid response was observed in 44.4% of patients at week 12, 30.8% at week 24, and 50% by the end of follow-up. Hemoglobin levels significantly improved at all time points, and adverse events were mild, affecting 16.7% of patients. The relapse rate was low, with only two patients relapsing and one progressing to acute myeloid leukemia. Luspatercept demonstrated good efficacy and safety in treating anemia in myelofibrosis patients.
https://doi.org/10.1007/s00277-024-05847-0
Autoimmune myelofibrosis: A Mayo Clinic series of 22 patients
In this study of 22 patients with autoimmune myelofibrosis (AIMF), 77% were female, with a median age of 45 years. Pancytopenia was present in 32%, and 59% required transfusions for anemia. Most patients (83%) had a history of autoimmune disease, and they were negative for JAK2, CALR, and MPL mutations. A complete response (CR), marked by resolution of cytopenias, was achieved in 74% of evaluable cases. First-line treatments included steroids alone or with immunosuppressants, cyclosporin, and mycophenolate, achieving CR in 54%, 50%, and 50% of cases, respectively. Rituximab as a salvage therapy was highly effective, with an 80% CR rate, offering a promising steroid-sparing option for AIMF
https://doi.org/10.1111/bjh.19499
Momelotinib as a safe and effective treatment option for cytopenic myelofibrosis patients
This real-world analysis of momelotinib in 60 myelofibrosis (MF) patients demonstrated significant improvements in anemia (84% hemoglobin rise), platelet counts (67%), and transfusion independence (21% within 4 weeks). Symptom relief occurred in 47% and spleen size reduced in 25% (median 6 weeks). Creatinine increases (17%) were manageable, and treatment discontinuation was rare (8% due to side effects). Momelotinib is effective and safe, even in heavily pre-treated cytopenic MF patients.
https://doi.org/10.1007/s00277-024-05908-4
Efficacy and safety of fedratinib in patients with myelofibrosis previously treated with ruxolitinib
The FREEDOM2 trial evaluated fedratinib versus best available therapy (BAT) in patients with myelofibrosis relapsed, refractory, or intolerant to ruxolitinib. Among 201 treated patients, spleen volume reduction (SVR) ≥35% by cycle 6 was achieved in 36% of fedratinib-treated patients compared to 6% in the BAT group. Fedratinib was associated with higher rates of grade ≥3 adverse events (40% vs. 12%), primarily anemia and thrombocytopenia, but gastrointestinal side effects were manageable with prophylactic antiemetics and thiamine supplementation. These findings support fedratinib as a viable second-line therapy for patients with myelofibrosis post-ruxolitinib, with effective strategies to mitigate adverse effects.
https://doi.org/10.1016/S2352-3026(24)00212-6
An Initiative of
Veenadhare Edutech Private Limited
1299, 2nd Floor, Shanta Nivas,
Beside Hotel Swan Inn, Off J.M.Road, Shivajinagar
Pune - 411005
Maharashtra – India

howitreat.in
CIN: U85190PN2022PTC210569
Email: admin@howitreat.in
Disclaimer: Information provided on this website is only for medical education purposes and not intended as medical advice. Although authors have made every effort to provide up-to-date information, the recommendations should not be considered standard of care. Responsibility for patient care resides with the doctors on the basis of their professional license, experience, and knowledge of the individual patient. For full prescribing information, including indications, contraindications, warnings, precautions, and adverse effects, please refer to the approved product label. Neither the authors nor publisher shall be liable or responsible for any loss or adverse effects allegedly arising from any information or suggestion on this website. This website is written for use of healthcare professionals only; hence person other than healthcare workers is advised to refrain from reading the content of this website.