
howitreat.in
A user-friendly, frequently updated reference guide that aligns with international guidelines and protocols.
Mature T-cell and NK-cell leukaemias (T-PLL, LGL Leukemia, ATLL)
This category includes following disorders:
- T-prolymphocytic leukaemia
- T-large granular lymphocytic leukaemia
- NK-large granular lymphocytic leukaemia
- Adult T-cell leukaemia/lymphoma
- Aggressive NK-cell leukaemia
- Sezary syndrome (Discussed under Mycosis fungoides)
T-Prolymphocytic Leukaemia
Introduction:
- Clonal proliferation of prolymphocytes with a mature post-thymic T-cell phenotype
- Associated with juxtaposition of TCL1A or MTCP1 gene next to a TCR locus.
Epidemiology:
- Account for 2% cases of mature lymphoid leukemias
- Median age 65 years
- Common in males
Etiopathogenesis:
inv(14)(q11q32) or t(14;14)(q11;q32)
↓
Juxtapositioning of TCL1A to the TCR gene enhancer loci of TRA/TRD
↓
Constitutive expressions of these TCL1 oncogene
↓
Enhanced AKT signaling
↓
Increased cell proliferation and survival.
Clinical Features:
- Skin lesions seen in 1/3 rd patients- Localised erythema in face and ear, nodules and erythroderma producing non-scaling, papular and non-pruritic rash.
- Fatigue, weakness, weight loss
- Hepatosplenomegaly
- Lymphadenopathy
- Effusions in 15%
Investigations:
- Hemogram:
- Anemia
- Thrombocytopenia
- Lymphocytosis with abnormal lymphoid cells (Usually >1lac/cmm)- Small to medium sized cells with round / Oval / markedly irregular nuclei. Condensed chromatin with visible nucleoli present. Cytoplasm is non granular and basophilic. Cytoplasmic protrusions / blebs are seen
- BM aspiration and biopsy– Diffuse infiltration of same type of cells, interstitial infiltrate
- Lymph node biopsy-
- Diffuse infiltration of tumor cells with predominant paracortical involvement
- Prominent, numerous high endothelial venules which are infiltrated by tumor cells
- Immunophenotyping
- Positive - CD2, 3, 7, CD52, CD5, TCP alpha-beta
- Negative- TdT, CD1a, B cell markers, HLA-DR, CD34, TCR-gamma-delta, CD10, CD11c, CD25, CD56, CD117
- CD4/CD8 expression varies
- Serology for HTLV I/II negative
- LFT- Impaired
- Uric acid, LDH- increased
- Molecular studies-
- T-cell antigen receptor chains are clonally arranged
- Deletions/ mutations of ATM genes may be present.
- Cytogenetics
- Abnormalities involving chromosome 14
- 80% have inv (14) with break points of q11 & q32
- 10% have t (14:14) (q11: q32)
- These translocations juxtapose TCR locus with the oncogene TCL1 on 14q32
- Other abnormalities- idic (8p11), t (8:8), trisomy 8q, del 12p13, t(x: 14)
- Abnormalities involving chromosome 14
Criteria for diagnosis:
Essential (Major) criteria:
- Peripheral blood lymphocytosis >5000/cmm or bone marrow infiltrate with T-PLL immunophenotype
- T-cell monoclonality
- TCL1A or MTCP1 rearrangement, alternatively TCL1A protein expression
Minor diagnostic criteria (At least 1 required)
- Abnormalities involving chromosome 11 (11q22.3, ATM)
- Abnormalities in chromosome 8: idic(8)(p11), t(8;8), trisomy 8q
- Abnormalities in chromosome 5, 12, 13, or 22, or complex karyotype
- Involvement of specific sites (e.g. splenomegaly, effusions)
Prognosis:
- Median survival <2 years
- Poor prognosis with
- Over-expression of TCL1 and MTCP1
- Age above 65 years
- Presence of serous effusions
- Hepatic or CNS involvement
- Bulky lymphadenopathy
- Very high absolute lymphocyte counts
- Complex karyotype
- Bone marrow suppression
- Organ dysfunction.
Differential Diagnosis:
- Polyclonal T lymphocytosis
- Large granular lymphocytic leukemia
- Adult T cell leukemia/ lymphoma
- Sezary syndrome
Pretreatment Work-up:
- History
- B-Symptoms
- Examination
- LN:
- Spleen:
- WHO P. S.
- BSA
- IHC/Flow cytometry
- BMA and Bx
- CT (CAP) or PET/CT scan
- Stage
- Hemoglobin
- TLC, DLC
- Platelet count
- LFT- Bili- T/D SGPT: SGOT: Albumin: Globulin:
- Creatinine
- Electrolytes: Na: K: Ca: Mg: PO4:
- Uric acid:
- LDH
- HIV:
- HBsAg:
- HCV:
- HTLV 1 Serology (Do western blot if ELISA is +ve)
- CMV Serology (If Alemtuzumab planned)
- UPT
- Cytogenetics
- FISH for inv (14), t(14:14), trisomy 8
- ECHO (If anthracyclines planned)- LVEF- %
- Chemotherapy consent after informing about disease, prognosis, cost of therapy, side effects, hygiene, food and contraception
- Fertility preservation
- PICC line insertion and Chest X ray after line insertion
- Tumor board meeting and decision
- Attach supportive care drug sheet
- Inform primary care physician
Treatment Plan:

T-Large Granular Lymphocytic Leukaemia
Introduction:
- It is a heterogeneous disorder characterized by persistent (>6 months) increase in the number of peripheral large granular lymphocytes, usually between 2,000-20,000/cmm, without a clearly identifiable cause.
Epidemiology:
- Represent 2-3% of cases of SLL
- Median age- 55 years
- M:F= 1:1
Etiology:
- Seen in patients with auto immune disorders (RA, SLE, Sjogren, Feltry’s syndrome)
- Has HLA DR4 association
- Sometimes associated with PNH, MDS and AML.
- Somatic mutation of STAT3 is seen in 1/3rd of patients.
Pathogenesis:
Sustained immune stimulation
↓
Activation of prosurvival pathways and hence cells fail to undergo apoptosis
↓
Increased IL15, which leads to increased survival of NK and T cells
↓
Release of soluble agents such as FasL and perforin by tumor cells
↓
Cytopenia.
Clinical Features:
- Usually asymptomatic
- Cytopenias
- Usually neutropenia- Due to antineutrophil antibody dependent cytotoxicity, maturation arrest mediated by neoplastic cells, enhanced FAS dependent apoptosis
- Thrombocytopenia- Due to inhibition of megakaryocytes, immune destruction of platelets and splenic sequestration.
- Autoimmune haemolytic anemia
- Auto immune disease
- B Symptoms
- Pure red cell aplasia- Seen in 15% patients
- Splenomegaly
- Lymphadenopathy
- Hepatomegaly
- Skin rash (rarely)
- Pulmonary hypertension- Due to lysis of endothelial cells resulting from activation of NK receptors via signaling partners DAP 10 and DAP12)
Investigations:
- Hemogram
- Lymphocyte- count is normal/ increased but majority of cells are large granular lymphocytes
- Large Granular Lymphocytes are large cells. Cytoplasm is abundant with fine / coarse azurophillic granules. Nucleus is eccentric with mature chromatin. No visible nucleolus
- LGL Count: 2-20 x 109 /L (usually > 5 x 109 /L)
- Neutropenia
- Sometimes anemia and thrombocytopenia
- BM aspiration and biopsy
- Normocellular or hypocellular in 50% patients, in rest of patients- hypercellular.
- Mild lymphoid infiltration in interstitial pattern
- Some times erythroid and myeloid hyperplasia with maturation arrest is seen
- Granulocytes show shift to left
- ANA, ANCA- may be positive
- S. protein electrophoresis: Hyper / Hypo gammaglobulinaemia
- Immunophenotyping
- Positive – CD2, CD3, CD8, TIA – 1, CD16, CD5, CD7, CD16, CD57, Cytotoxic proteins (TIA1, Granzyme B)
- Negative –CD56, CD4, CD7, TCP gamma-delta
- Variable – CD11b, CD56, CD57
- Molecular studies: T-cell receptor gene rearrangements. (Note: Clonal TCR rearrangement without cytologic or immunophenotypic evidence of abnormal T cell population does not constitute a diagnosis of T cell malignancy)
- Cytogenetic – Nothing specific
Criteria for Diagnosis: (All 3 essential criteria or 2 essential criteria+1 desirable citeria must be present for diagnosis)
Essential
- An increase in circulating cytotoxic T-cells, often greater than 2000/cmm but may be less
- Presence of a T-cell population (usually CD8 positive) with down-regulation of CD5 and/or CD7 and/or abnormal expression of CD16 and NK-cell associated receptors
- Evidence of T-cell monoclonality
Desirable:
- Bone marrow immunohistochemistry revealing intra-sinusoidal cytotoxic lymphocyte infiltrates
- Demonstration of STAT3 or STAT5B mutation
Prognosis:
- Indolent course
- 80% survive at 4 years,
- Median survival > 10 years
Differential Diagnosis:
- Clonal reactive lymphocytosis
- Count is usually < 5 x 109 / L
- TCR gene rearrangements in germ line configuration
- Seen following allogeneic stem cell transplantation, B-NHL, CML patients on Imatinib
- Chronic neutropenia
- PRCA
- Rhematoid arthritis
- HIV infection
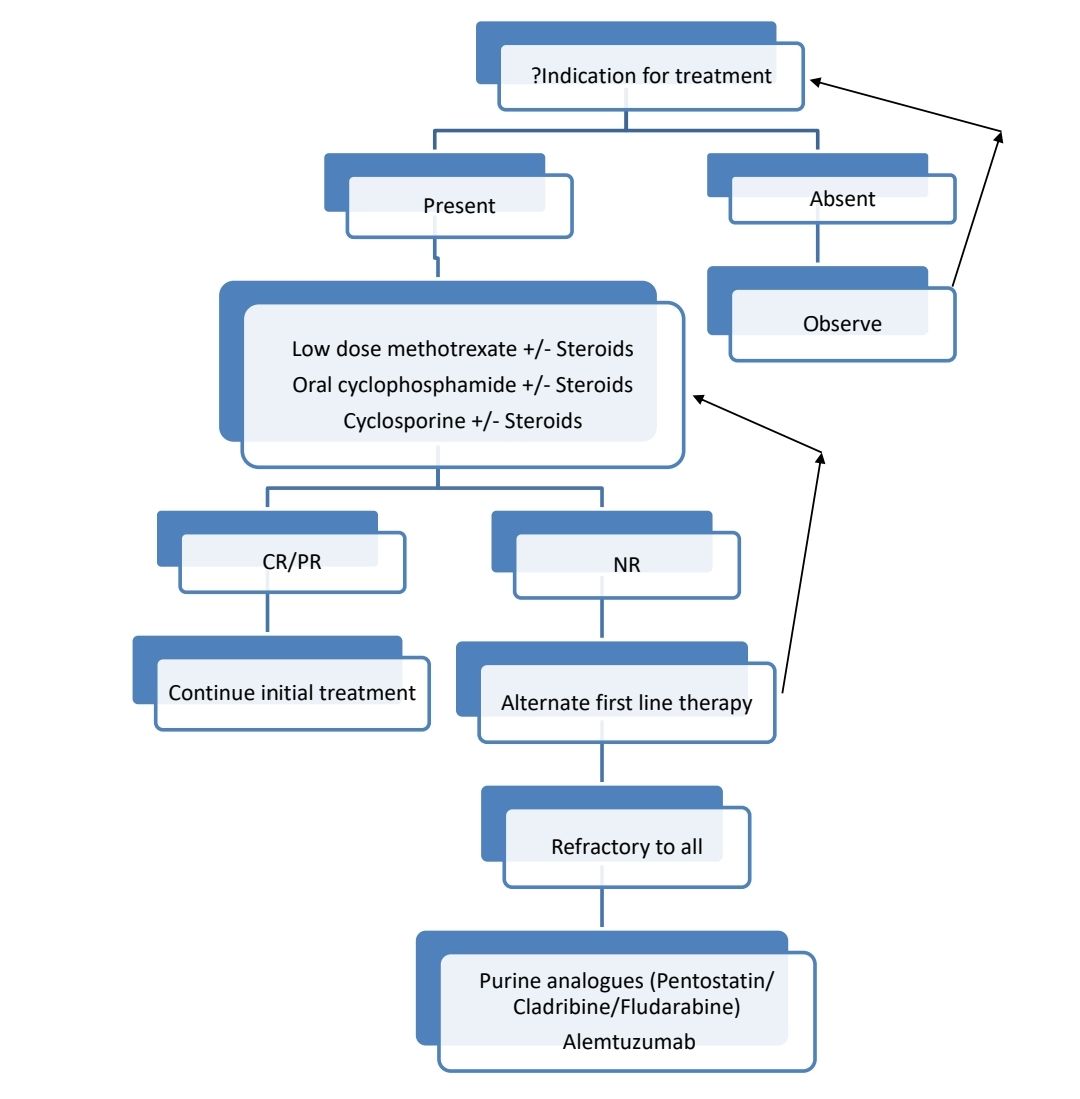
Indications for Treatment:
- Severe cytopenias (ANC <500/cmm or Hemoglobin <10gm/dL, or Platelet count <50,000/cmm)
- Autoimmune diseases with T-LGL requiring therapy
- Severe B symptoms
- Pulmonary artery hypertension secondary to LGL
- Recurrent infections
- Progressive lymphocytosis / organomegaly
Pretreatment Work-up:
- History
- B-Symptoms
- Autoimmune diseases especially rheumatoid arthritis
- Examination
- LN:
- Spleen:
- WHO P. S.
- BSA
- Flow cytometry
- CT (CAP)
- Hemoglobin
- TLC, DLC
- Platelet count
- LFT- Bili- T/D SGPT: SGOT: Albumin: Globulin:
- Creatinine
- Electrolytes: Na: K: Ca: Mg: PO4:
- Uric acid:
- LDH
- HIV:
- HBsAg:
- HCV:
- Serology for HTLV- 1, 2
- PCR for viral DNA- CMV(If alemtuzumab is planned)
- Screen for autoimmune diseases
- RA Factor: ANA: ESR:
- UPT
- STAT 3 and STAT5B Mutation tests
- ECHO (If anthracyclines planned/RHF)- LVEF- %
- Chemotherapy consent after informing about disease, prognosis, cost of therapy, side effects, hygiene, food and contraception
- Fertility preservation
- PICC line insertion and Chest X ray after line insertion
- Tumor board meeting and decision
- Attach supportive care drug sheet
- Inform primary care physician
Treatment plan:
Response Criteria: Response assessment should be done after 4 months of treatment
- Complete response:
- Recovery of counts with Hemoglobin >12gm/dL, ANC >1500/cmm, Platelet count >1.5lac/cmm
- Resolution of lymphocytosis (<4000/cmm)
- Circulating LGL- <500/cmm
- Partial response:
- Recovery of counts, Hb>8gm/dL, ANC >500/cmm, Platelet count- >50,000/cmm
About Each Modality of Treatment:
- Cyclosporin A
- 5-10mg/kg/day in 2 divided doses.
- Must be titrated to achieve response.
- Enhanced response with use of G-CSF or EPO.
- 3-4 months treatment is required to achieve response.
- Responders need long term maintenance therapy.
- Goal of treatment is correction of cytopenia.
- Methotrexate
- 10 mg/m2 – weekly pulse- Tapered after 1 month of therapy
- Cyclophosphamide
- 200 – 300 mg / week.
- Limit use of cyclophosphamide to 4 months if no response and to less than 12 months, if PR is observed at 4 months, due to increased risk of leukemogenesis.
- Corticosteroids
- Prednisolone- 1mg/Kg
Other Treatment Options: Newer treatment options:
- Tipifarnib- Farnesyltransferase inhibitor
- STAT3 inhibitors
- Bortezomib
- PI3K inhibitors
- Anti CD2- Siplizumab
- Anti CD112 Antibodies
- Anti- IL15
- Monoclonal antibody against FAS ligand
- Imatinib
NK-Large Granular Lymphocytic Leukaemia
- Neoplasm characterized by a persistent increase in peripheral blood NK cells (usually greater than 2000/cmm) in the absence of a clearly identifiable cause
- Mutations of STAT3, CCL22 and TET2 are seen
- Immunophenotyping- Positive for CD2, CD7, CD16, CD56, CD57, cytotoxic granule proteins (TIA-1, Granzyme B, Granzyme M) and negative for CD3, CD4, CD8 and CD5
- Compared to T-LGL
- Seen in younger patients
- High incidence of B symptoms
- More massive hepatosplenomegaly
- Lymphadenopathy
- GI involvement is common
- Criteria for diagnosis:
- Essential:
- An increase in circulating NK-cells, typically greater than 2000/cmm, persisting greater than 6 months
- Flow cytometric evidence of peripheral blood or bone marrow aspirate involvement by a uniform population of surface CD3 negative, CD16 positive NK-cell population.
- A KIRs restricted pattern of expression, demonstrated by flow cytometry analysis (either a dominant expression of a relevant KIR or lack of them), is accepted as a surrogate marker of clonal expansion
- Essential:
- Desirable:
- Bone marrow involved by intra-sinusoidal cytotoxic CD8 positive NK-cells
- Demonstration of STAT3 and/or TET2 mutations with NK-cell lineage confirmed by flow cytometry
- If both 2 and 3 essential criteria are present, a diagnosis of NK-LGLL can be made in the absence of documented persistence of an absolute peripheral blood NK-cell count of greater than 2000/cmm.
- Prognosis: Indolent course
- Treatment: Similar to T-LGL leukemia
Adult T-cell Leukaemia/Lymphoma
Epidemiology:
- Common in Japan, Caribbean, Africa, and South America.
- Median age- 62 years
Etiopathogenesis:
- HTLV 1- P40 tax viral protein causes transcriptional activation of many genes in infected lymphocytes
- HTLV1-Basic leucine zipper factor causes T cell proliferation and oncogenesis
- Lifetime risk of developing leukemia in infected patients is 1-5%.
Clinical Features: There are 4 variants of this disease
- Acute variant (Leukemia variant)
- Increased WBC count
- Skin rash
- Generalized lymphadenopathy
- Hypercalcemia with / without lytic bone lesions
- Hepatosplenomegaly
- CNS involvement- Seen in 10% patients
- B Symptoms
- Opportunistic infections- PCP, Aspergillus, Candida, strongyloidiasis, CMV
- Lymphomatous variant
- Prominent lymphadenopathy without peripheral blood involvement
- Chronic variant
- Skin lesions – exfoliative rash
- Absolute lymphocytosis (>4000/cmm) with T lymphocytes (>3500/cmm)
- Smoldering variant
- Normal WBC count without <5% circulating neoplastic cells.
Investigations:
- Peripheral smear:
- Neoplastic lymphoid cells are medium to large sized and contain pleomorphic nuclei. Chromatin is coarsely clumped and there is distinct nucleoli.
- Some cells contain polylobated/ convoluted/ cerebriform nuclei (hence called flower cells/ cloverleaf cells)
- Cytoplasm is deeply basophilic
- Small proportion of blast like cells are present
- Eosinophilia may be present
- Anemia and thrombocytopenia
- Bone marrow aspiration and biopsy: Patchy infiltrate of tumor cells
- Skin biopsy- Poutrier like micro abscesses
- Lymph node biopsy
- Leukemia pattern of infiltration
- Dilation of LN sinuses
- Immunophenotyping
- Positive- Mature T-cell associated antigens (CD-2, 3, 5), CD4, CD30, CD25, TCR Alfa-beta, CCR4, FOXP3, CD29, HLA-DR
- Negative – CD7, 8, NK cell markers, CD11b, CD11c, TIA-1, Granzyme
- Any CD4 and CD8 combinations are possible.
- Molecular studies:
- Clonally integrated HTLV-1 is found
- TCR genes clonally rearranged
- LDH – Increased
- LFT – Abnormal
- Serum calcium level- Elevated (Due to release of cytokines like parathyroid like hormone, IL-1& TNF- beta)
- Serology for HTLV antibodies- Positive
Criteria for Diagnosis:
Essential:
- Neoplastic lymphoid cell proliferation with mature T-cell phenotype
- Proven HTLV-1 carriership
Desirable:
- Lymphoid cells with prominent convolutions and lobulation
- Identification of monoclonal integration of HTLV-1
Prognosis:
- Average survival – 5 months- 13 months (Poor with acute and lymphoma variants)
- Poor prognostic markers:
- Increased LDH
- Leucocytosis
- Hypercalcemia
- Age >40 years
- >3 involved lesions
- Poor performance score
- Thrombocytopenia
- Eosinophilia
- Bone marrow involvement
- CCR4 expression
- TP53 mutation.
Pretreatment Work-up:
- History
- B-Symptoms
- Examination
- LN:
- Spleen:
- WHO P. S.
- BSA
- IHC/Flow cytometry
- CT (CAP) or PET-CT
- Hemoglobin
- TLC, DLC
- Absolute number of atypical cells
- Platelet count
- LFT: Bili- T/D SGPT: SGOT: Albumin: Globulin:
- Creatinine
- Electrolytes: Na: K: Ca: Mg: PO4:
- Uric acid:
- LDH
- HIV:
- HBsAg:
- HCV:
- HTLV 1 (Do Western blot/PCR if serology is +ve)
- UPT
- Stool for Strongyloides
- CT/ MRI Brain (If symptomatic)
- CSF (in acute/ lymphoma subtypes and CNS symptoms)
- ECHO (If anthracyclines planned) LVEF- %
- Chemotherapy consent after informing about disease, prognosis, cost of therapy, side effects, hygiene, food and contraception
- Fertility preservation
- PICC line insertion and Chest X ray after line insertion
- Tumor board meeting and decision
- Attach supportive care drug sheet
- Inform primary care physician
Treatment Plan:
Exclude co-infection with Strongyloidiasis prior to therapy, as chemotherapy can lead to aggravation of infection.
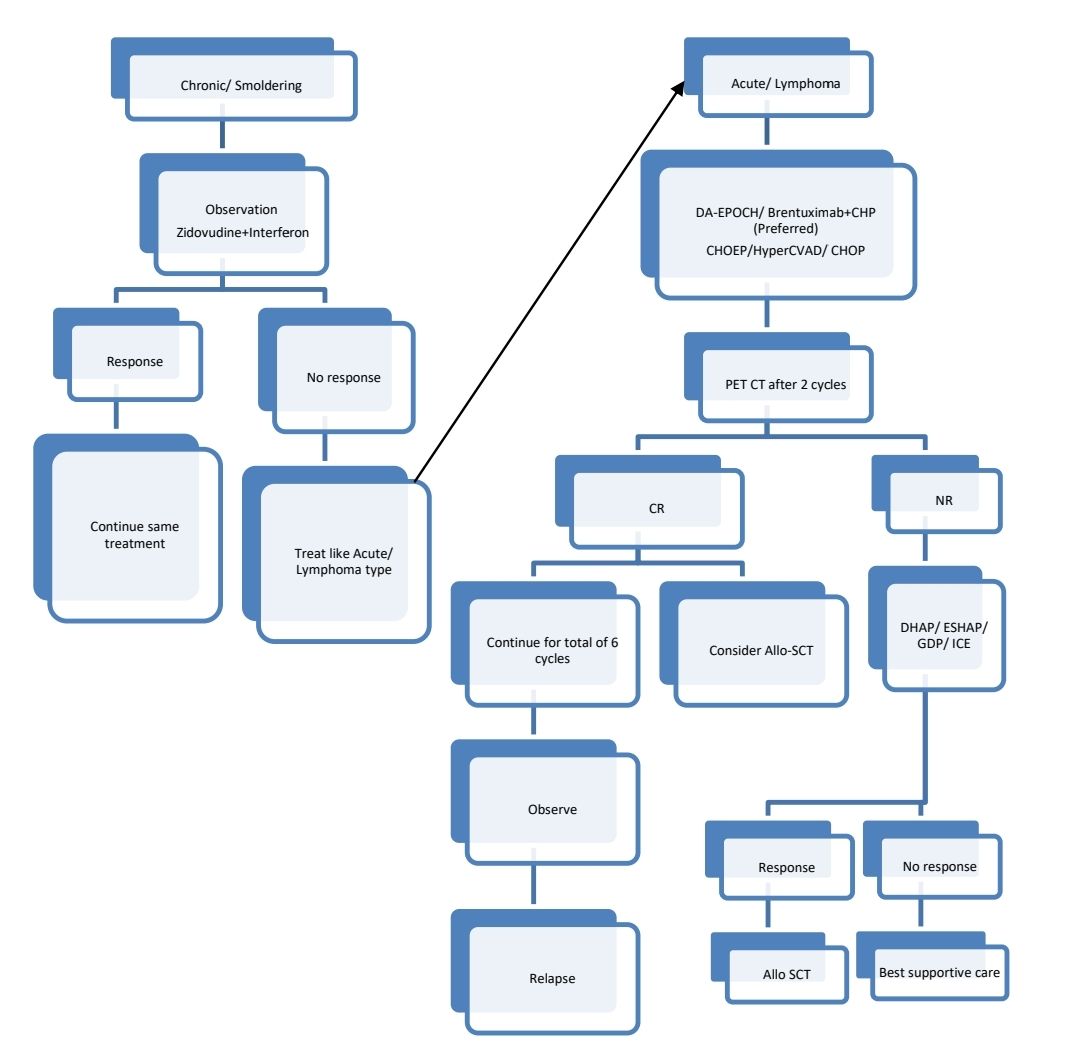
CNS prophylaxis with triple IT should be given.
Response Criteria:
- Complete response- All of the following
- Normal lymph nodes
- No extranodal masses
- No organomegaly
- Normal skin
- Normal peripheral blood
- Normal bone marrow
About Each Modality of Treatment:
- Interferon- 3 million units- OD + Zidovudine- 250mg- BD
- Both should be continued for 5 years if patient tolerates
Aggressive NK-cell Leukaemia
Introduction:
- It is a rare leukemic form of an NK cell neoplasm with aggressive clinical course.
- Generally skin and nose are not involved.
Epidemiology:
- Median age 40 years
- Rare tumor
- Common in Asia
- Median age- 39 years
Etiology:
- EBV infection
Clinical Features:Acute onset of
- Cytopenia
- B symptoms- Particularly fever
- Jaundice
- Lymphadenopathy
- Hepatosplenomegaly
- Sometimes
- DIC
- Hemophagocytic syndrome
- Liver dysfunction
- Multi-organ failure
Investigations:
- Peripheral smear
- Leukemic cells are slightly larger than normal large granular lymphocytes. Nucleus is normal looking or enlarged, with irregular folding and open chromatin. Nucleoli are inconspicuous / distinct. Cytoplasm is abundant and lightly basophilic
- Bone marrow aspiration and biopsy
- Massive/ subtle/ focal infiltrate of tumor cells
- Reactive histiocytes with hemophagocytosis
- Necrosis is common
- Immunophenotyping
- Positive – CD2, CD3E, CD7,CD56, CD16, Cytotoxic molecules
- Negative – CD3, CD5, CD8, CD57
- Molecular studies- TCR genes are in germ line configuration
- Cytogenetics- del(6) (q21 q25), del 11q, del 17p, i(7)
- S. LDH- Elevated
Criteria for diagnosis:
Essential
- Presentation with fever, constitutional symptoms and a leukemic blood picture
- Systemic (multi-organ) proliferation of neoplastic lymphoid cells with an NK-cell immunophenotype
- Lack of TCR protein expression and/or lack of clonal rearrangement of TR genes
Desirable:
- EBER positivity (positive in ~90% of cases)
- Haemophagocytic lymphohistiocytosis
Prognosis:
- Fatal outcome within 1 year
- Overall survival- 2 months
Treatment Plan:
- Asparginase based intensive chemotherapy (Intensive ALL type therapy) with CNS prophylaxis
- Consolidation with Allo SCT or HDT with ASCR in CR1.
Recent advances:
Effective treatment with the selective cytokine inhibitor BNZ-1 reveals the cytokine dependency of T-LGL leukemia
A phase 1/2 clinical trial of BNZ-1, a pegylated peptide that selectively inhibits the binding of interleukin-15 (IL-15) and other γc cytokines to their cellular receptor complex, was conducted in patients with T-cell large granular lymphocytic leukemia (T-LGLL) who had hematocytopenias. Clinical responses were assessed based on hematologic parameters, and BNZ-1 demonstrated clinical partial responses in 20% of T-LGLL patients with minimal toxicity. The study provides first-in-human proof that T-LGL leukemic cells are dependent on IL-15, and intervention with BNZ-1 shows therapeutic effects, shedding light on the pathogenesis of this disease.
https://doi.org/10.1182/blood.2022017643
Valemetostat for relapsed or refractory adult T-cell leukemia/lymphoma
Valemetostat is a potent enhancer of zeste homolog 2 (EZH2) and EZH1 inhibitor. Patients received valemetostat 200 mg/day orally until progressive disease or unacceptable toxicity. Valemetostat demonstrated promising efficacy and tolerability in heavily pretreated patients, warranting further investigation in treating R/R ATL.
https://doi.org/10.1182/blood.2022016862
First-line mogamulizumab-containing chemotherapy in adult T-cell leukaemia-lymphoma
Mogamulizumab is a humanized monoclonal antibody directed against C-C chemokine receptor 4. Present study retrospectively analysed clinical outcomes in 39 transplant-ineligible patients with untreated aggressive ATL at Kumamoto University Hospital between 2010 and 2021. The probability of four-year overall survival was 46.3% in the first-line Mog-containing treatment group compared to 20.6% in the chemotherapy-alone group.
https://doi.org/10.1111/bjh.18281
Ruxolitinib in the treatment of relapsed/refractory large granular lymphocytic leukaemia
Large granular lymphocytic (LGL) leukemia is a rare disorder characterized by cytotoxic T or NK cell expansion. For patients with relapsed or refractory disease, ruxolitinib—a JAK/STAT pathway inhibitor—was evaluated in 21 patients, showing an 86% overall response rate, with 3 complete and 15 partial responses. 1-year event-free and overall survival rates were 57% and 83%, respectively. Ruxolitinib improved cytopenias with mild side effects, supporting its potential as a treatment for challenging cases of LGL leukemia.
https://doi.org/10.1111/bjh.19476
An Initiative of
Veenadhare Edutech Private Limited
1299, 2nd Floor, Shanta Nivas,
Beside Hotel Swan Inn, Off J.M.Road, Shivajinagar
Pune - 411005
Maharashtra – India

howitreat.in
CIN: U85190PN2022PTC210569
Email: admin@howitreat.in
Disclaimer: Information provided on this website is only for medical education purposes and not intended as medical advice. Although authors have made every effort to provide up-to-date information, the recommendations should not be considered standard of care. Responsibility for patient care resides with the doctors on the basis of their professional license, experience, and knowledge of the individual patient. For full prescribing information, including indications, contraindications, warnings, precautions, and adverse effects, please refer to the approved product label. Neither the authors nor publisher shall be liable or responsible for any loss or adverse effects allegedly arising from any information or suggestion on this website. This website is written for use of healthcare professionals only; hence person other than healthcare workers is advised to refrain from reading the content of this website.